Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
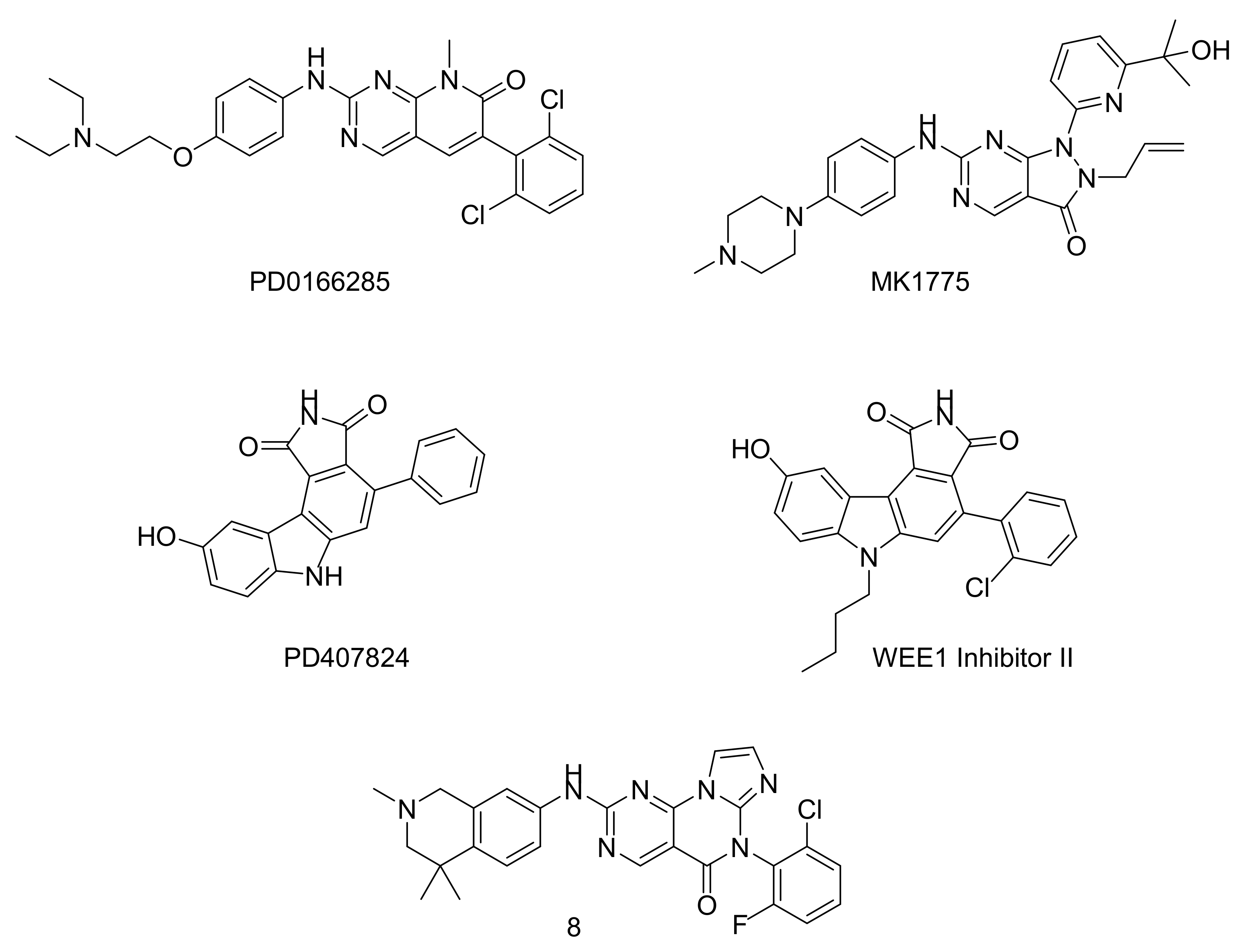{kind=link}
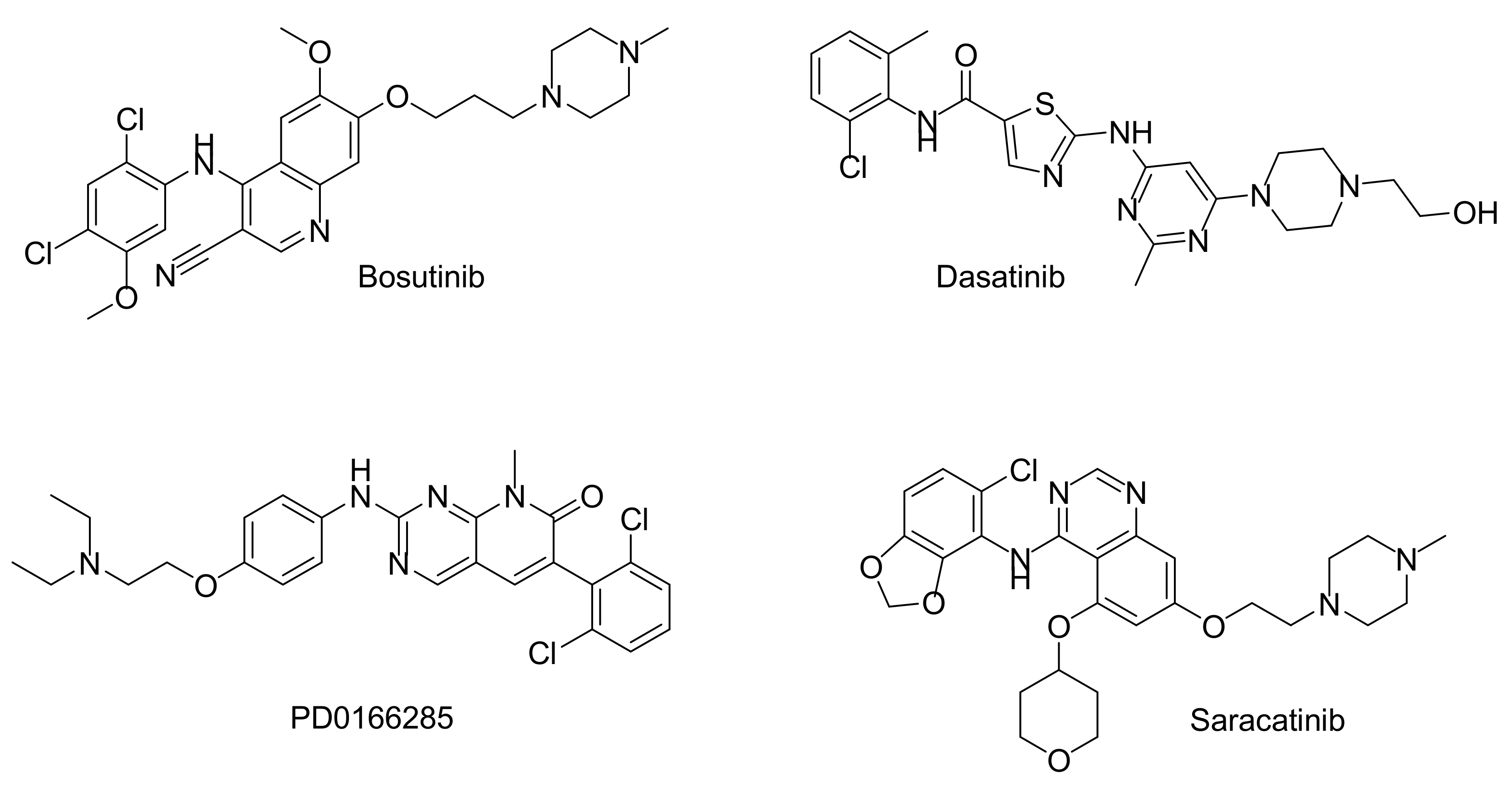{kind=link}
Abstract
:1. Introduction
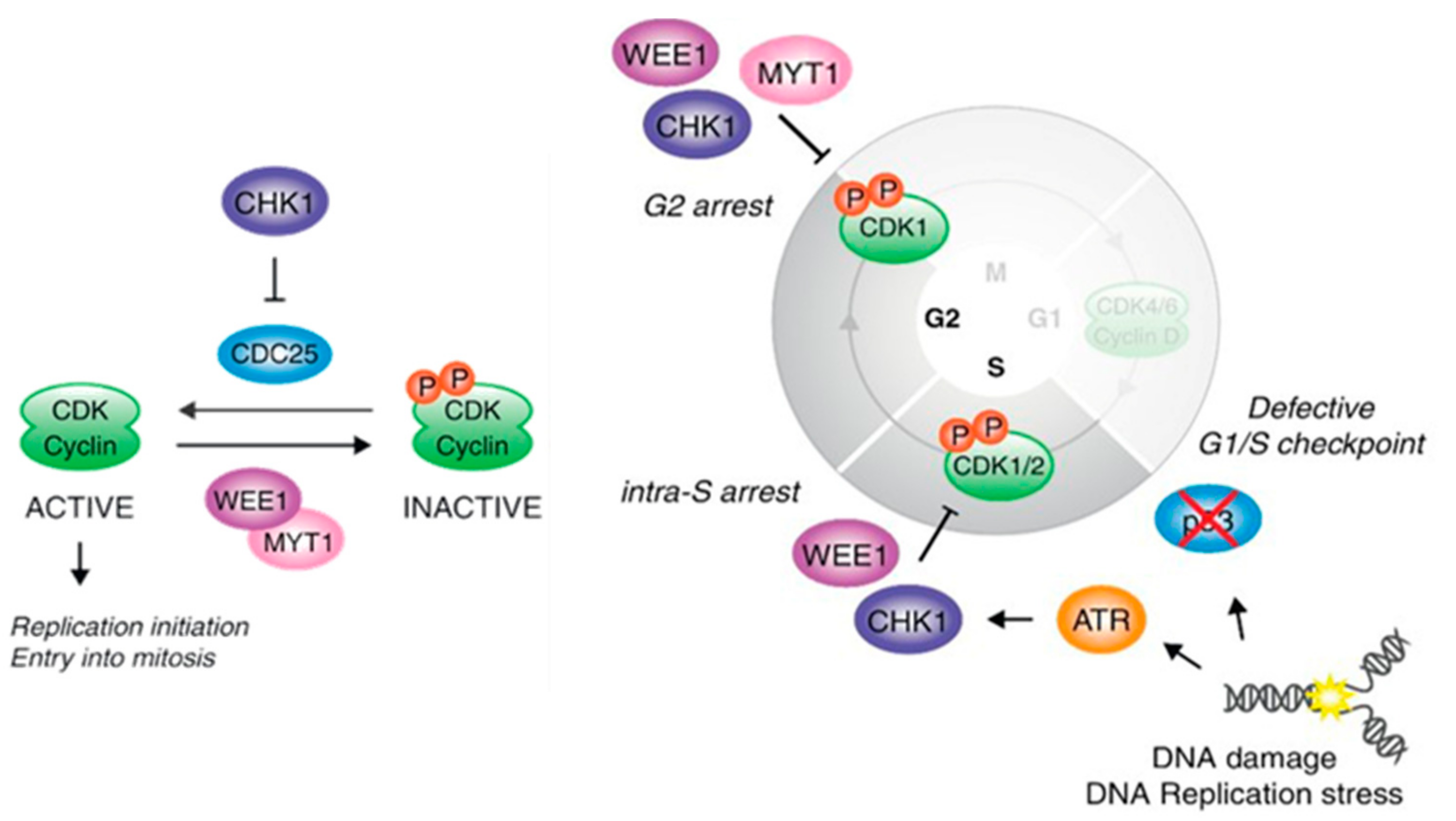
2. Physiological Role of WEE Family Kinases
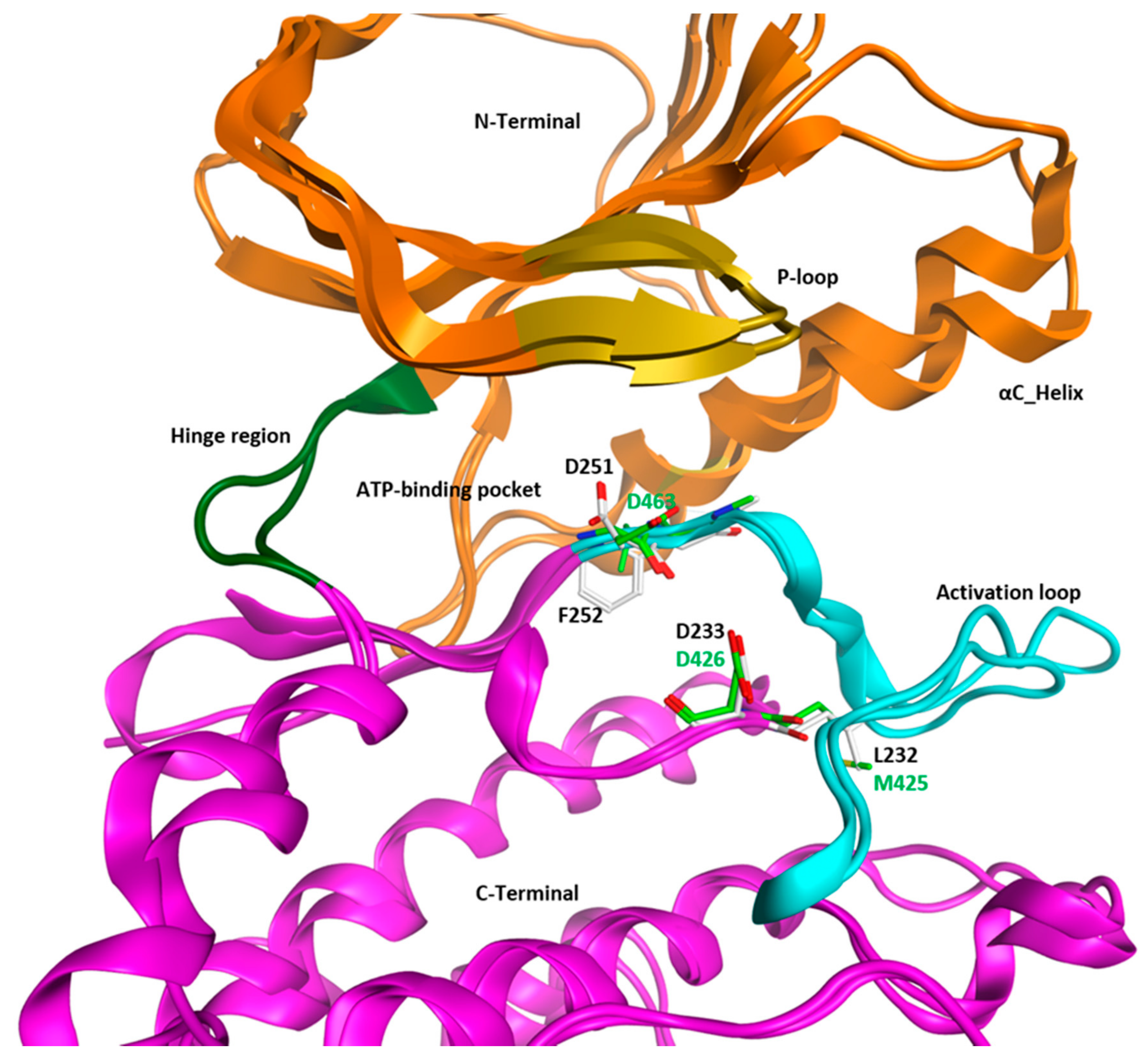
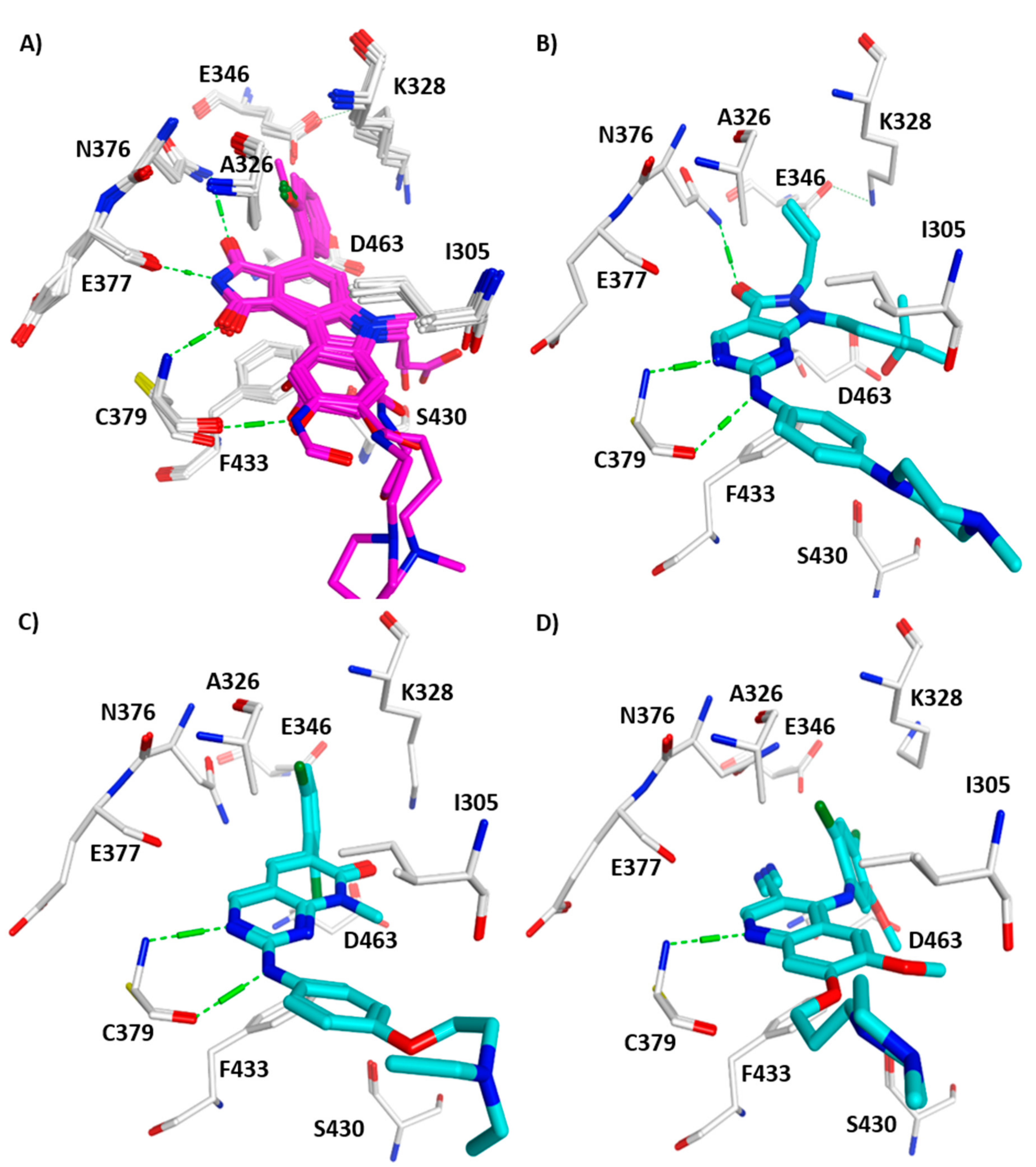
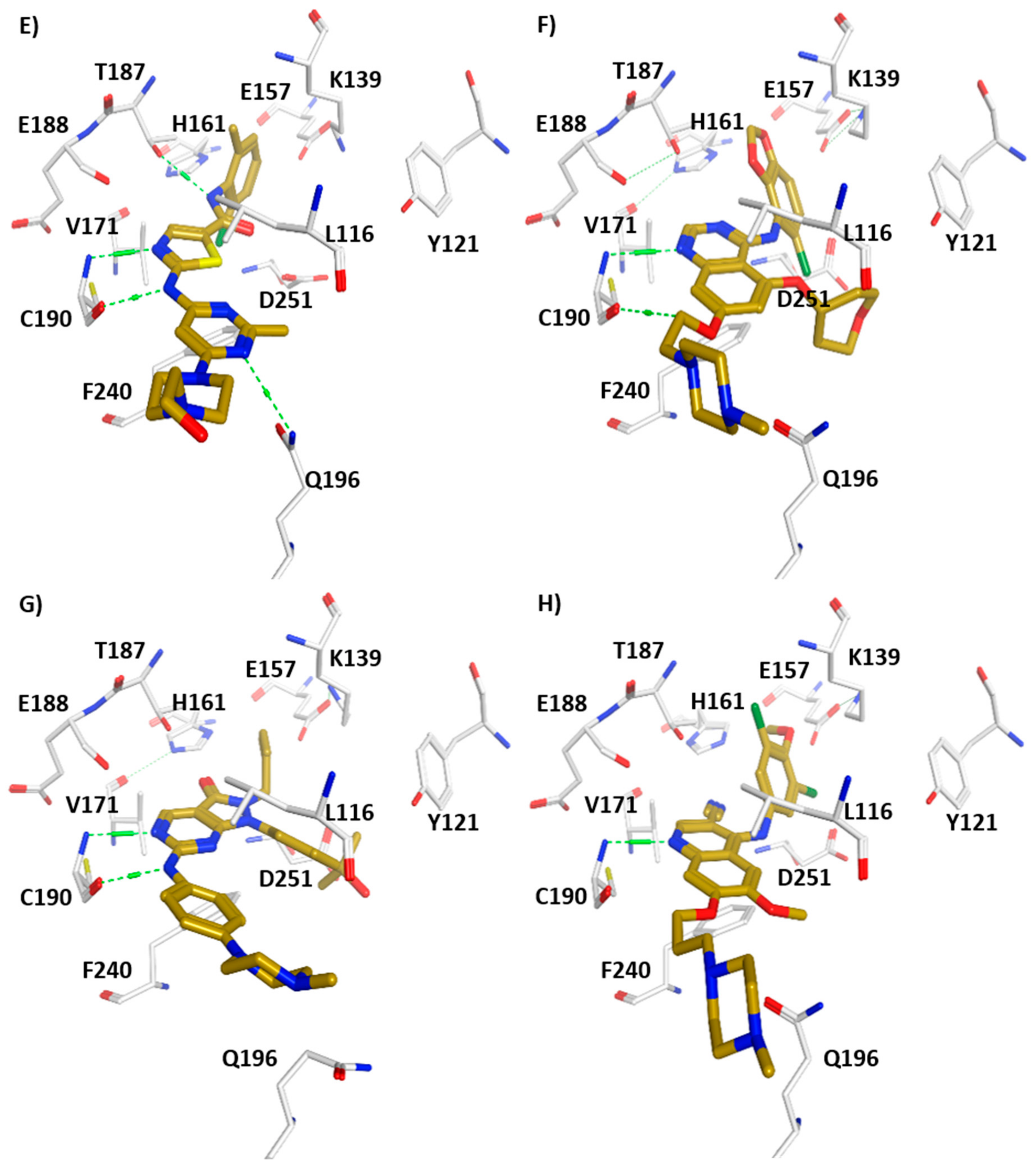
3. Structural Analysis of WEE Family Kinases
4. WEE1 and PKMYT1 as Potential Drug Targets in Cancer Therapy
4.1. Assays
4.2. Cell Experiments
4.3. Clinical Trials with PKMYT1 and WEE1 Inhibitors
5. Summary and Perspectives
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Hunter, T. Protein kinase signaling networks in cancer. Curr. Opin. Genet. Dev. 2011, 21, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Sananes, M.R.; Kapuy, O.; Hunt, T.; Novak, B. Switches and latches: A biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3584–3594. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Rodriguez-Bravo, V.; Medema, R.H. The decision to enter mitosis: Feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009, 185, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Pomerening, J.R. Positive-feedback loops in cell cycle progression. FEBS Lett. 2009, 583, 3388–3396. [Google Scholar] [CrossRef] [PubMed]
- Wolanin, P.M.; Thomason, P.A.; Stock, J.B. Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Vulpetti, A.; Bosotti, R. Sequence and structural analysis of kinase ATP pocket residues. Farmaco 2004, 59, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M.; Reisine, T. Human kinome drug discovery and the emerging importance of atypical allosteric inhibitors. Expert Opin. Drug Discov. 2010, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Ando, H.; Watanabe, N.; Kitamura, K.; Ito, K.; Okayama, H.; Miyamoto, T.; Agui, T.; Sasaki, M. Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells 2000, 5, 839–847. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [PubMed]
- Mueller, P.R.; Coleman, T.R.; Kumagai, A.; Dunphy, W.G. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 1995, 270, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Roche, V.F. Cancer and Chemotherapy. In Foye’s Principles of Medicinal Chemistry; Lemke, T., Williams, D.A., Eds.; Lippincott, Williams & Wilkins: Baltimore, MD, USA, 2008; pp. 1147–1192. [Google Scholar]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Macurek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, A.; Kaldis, P. Mammalian cell-cycle regulation: Several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009, 28, 2925–2939. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Woodbury, E.L.; Quang, P.N.; Paraz, M.; Blethrow, J.D.; Shah, K.; Shokat, K.M.; Morgan, D.O. Targets of the cyclin-dependent kinase Cdk1. Nature 2003, 425, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Ptacek, J.; Devgan, G.; Michaud, G.; Zhu, H.; Zhu, X.; Fasolo, J.; Guo, H.; Jona, G.; Breitkreutz, A.; Sopko, R.; et al. Global analysis of protein phosphorylation in yeast. Nature 2005, 438, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.K.; Poon, R.Y. A roller coaster ride with the mitotic cyclins. Semin. Cell Dev. Biol. 2005, 16, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Pines, J.; Hunter, T. Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol. 1991, 115, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gautier, J.; Norbury, C.; Lohka, M.; Nurse, P.; Maller, J. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+. Cell 1988, 54, 433–439. [Google Scholar] [CrossRef]
- Lohka, M.J.; Hayes, M.K.; Maller, J.L. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 1988, 85, 3009–3013. [Google Scholar] [CrossRef] [PubMed]
- Gautier, J.; Minshull, J.; Lohka, M.; Glotzer, M.; Hunt, T.; Maller, J.L. Cyclin is a component of maturation-promoting factor from Xenopus. Cell 1990, 60, 487–494. [Google Scholar] [CrossRef]
- O’Farrell, P.H. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001, 11, 512–519. [Google Scholar] [CrossRef]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef] [PubMed]
- Booher, R.N.; Holman, P.S.; Fattaey, A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J. Biol. Chem. 1997, 272, 22300–22306. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Shinya, N.; Iwamatsu, A.; Nishida, E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001, 410, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Pomerening, J.R.; Ubersax, J.A.; Ferrell, J.E., Jr. Rapid cycling and precocious termination of G1 phase in cells expressing CDK1AF. Mol. Biol. Cell 2008, 19, 3426–3441. [Google Scholar] [CrossRef] [PubMed]
- Pomerening, J.R.; Sontag, E.D.; Ferrell, J.E., Jr. Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 2003, 5, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; van Zon, W.; Karlsson Rosenthal, C.; Wolthuis, R.M. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 2007, 5, e123. [Google Scholar] [CrossRef] [PubMed]
- Pomerening, J.R. Uncovering mechanisms of bistability in biological systems. Curr. Opin. Biotechnol. 2008, 19, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bravo, V.; Guaita-Esteruelas, S.; Salvador, N.; Bachs, O.; Agell, N. Different S/M checkpoint responses of tumor and non tumor cell lines to DNA replication inhibition. Cancer Res. 2007, 67, 11648–11656. [Google Scholar] [CrossRef] [PubMed]
- Jackman, M.; Lindon, C.; Nigg, E.A.; Pines, J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 2003, 5, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Jackman, M.; Kubota, Y.; den Elzen, N.; Hagting, A.; Pines, J. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol. Biol. Cell 2002, 13, 1030–1045. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.; Nurse, P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987, 49, 559–567. [Google Scholar] [CrossRef]
- Liu, F.; Stanton, J.J.; Wu, Z.; Piwnica-Worms, H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol. 1997, 17, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.R.; Leise, W.F. Measurement of Wee Kinase Activity. In Methods in Molecular Biology: Cell Cycle Control; Humphrey, T., Brooks, G., Eds.; Humana Press Inc.: New York, NY, USA, 2005; pp. 299–328. [Google Scholar]
- Tassan, J.P.; Schultz, S.J.; Bartek, J.; Nigg, E.A. Cell cycle analysis of the activity, subcellular localization, and subunit composition of human CAK (CDK-activating kinase). J. Cell Biol. 1994, 127, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.; Blow, J.; Nurse, P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991, 10, 3321–3329. [Google Scholar] [PubMed]
- Coulonval, K.; Kooken, H.; Roger, P.P. Coupling of T161 and T14 phosphorylations protects cyclin B-CDK1 from premature activation. Mol. Biol. Cell 2011, 22, 3971–3985. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol. 2010, 189, 247–259. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H.; Russell, P. Cell cycle regulation of human WEE1. EMBO J. 1995, 14, 2166–2175. [Google Scholar] [PubMed]
- Larochelle, S.; Merrick, K.A.; Terret, M.E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Wells, N.J.; Watanabe, N.; Tokusumi, T.; Jiang, W.; Verdecia, M.A.; Hunter, T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 1999, 112 Pt 19, 3361–3371. [Google Scholar] [PubMed]
- Wang, Y.; Decker, S.J.; Sebolt-Leopold, J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol. Ther. 2004, 3, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, J.; Scarpa, M.; Ortega-Bellido, M.; Malhotra, V. MEK1 inactivates Myt1 to regulate Golgi membrane fragmentation and mitotic entry in mammalian cells. EMBO J. 2013, 32, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Yonemura, S.; Murata, M.; Nakamura, N.; Piwnica-Worms, H.; Nishida, E. Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J. Cell Biol. 2008, 181, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Linardopoulos, S.; Turner, N.C. Tumour selective targeting of cell cycle kinases for cancer treatment. Curr. Opin. Pharmacol. 2013, 13, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Van Vugt, M.A.; Bras, A.; Medema, R.H. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol. Cell 2004, 15, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.P.; Poon, R.Y. The CDK1 inhibitory kinase MYT1 in DNA damage checkpoint recovery. Oncogene 2013, 32, 4778–4788. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Cuellar, R.A.; Berndt, N.; Lee, H.E.; Olesen, S.H.; Martin, M.P.; Jensen, J.T.; Georg, G.I.; Schonbrunn, E. Structural Basis of Wee Kinases Functionality and Inactivation by Diverse Small Molecule Inhibitors. J. Med. Chem. 2017, 60, 7863–7875. [Google Scholar] [CrossRef] [PubMed]
- Squire, C.J.; Dickson, J.M.; Ivanovic, I.; Baker, E.N. Structure and inhibition of the human cell cycle checkpoint kinase, Wee1A kinase: An atypical tyrosine kinase with a key role in CDK1 regulation. Structure 2005, 13, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Wichapong, K.; Rohe, A.; Platzer, C.; Slynko, I.; Erdmann, F.; Schmidt, M.; Sippl, W. Application of docking and QM/MM-GBSA rescoring to screen for novel Myt1 kinase inhibitors. J. Chem. Inf. Model. 2014, 54, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Noble, M.E.; Tucker, J.A. Cyclin-dependent kinases: Inhibition and substrate recognition. Curr. Opin. Struct. Biol. 1999, 9, 738–744. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Suganuma, M.; Kawabe, T.; Hori, H.; Funabiki, T.; Okamoto, T. Sensitization of cancer cells to DNA damage-induced cell death by specific cell cycle G2 checkpoint abrogation. Cancer Res. 1999, 59, 5887–5891. [Google Scholar] [PubMed]
- Dixon, H.; Norbury, C.J. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [Google Scholar] [PubMed]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Mante, S.; Minneman, K.P. Caffeine inhibits forskolin-stimulated cyclic AMP accumulation in rat brain. Eur. J. Pharmacol. 1990, 175, 203–205. [Google Scholar] [CrossRef]
- Cortez, D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J. Biol. Chem. 2003, 278, 37139–37145. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Valent, A.; Raslova, H.; Yakushijin, K.; Horne, D.; Feunteun, J.; Lenoir, G.; Medema, R.; et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 2004, 23, 4362–4370. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Mitotic arrest and cell fate: Why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007, 6, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updat. 2001, 4, 303–313. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Mir, S.E.; Noske, D.; Van Noorden, C.J.; Wurdinger, T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin. Cancer Res. 2011, 17, 4200–4207. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Gu, Y.; Morgan, D.O. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J. Cell Biol. 1996, 134, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, S. Checkpoint Kinase and Wee1 Inhibition as Anticancer Therapeutics. In DNA Repair in Cancer Therapy; Kelley, M.R., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 211–234. [Google Scholar]
- Tse, A.N.; Sheikh, T.N.; Alan, H.; Chou, T.C.; Schwartz, G.K. 90-kDa heat shock protein inhibition abrogates the topoisomerase I poison-induced G2/M checkpoint in p53-null tumor cells by depleting Chk1 and Wee1. Mol. Pharmacol. 2009, 75, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. Med. Chem. Commun. 2017, 8, 295–319. [Google Scholar] [CrossRef]
- Panek, R.L.; Lu, G.H.; Klutchko, S.R.; Batley, B.L.; Dahring, T.K.; Hamby, J.M.; Hallak, H.; Doherty, A.M.; Keiser, J.A. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther. 1997, 283, 1433–1444. [Google Scholar] [PubMed]
- Rohe, A.; Gollner, C.; Wichapong, K.; Erdmann, F.; Al-Mazaideh, G.M.; Sippl, W.; Schmidt, M. Evaluation of potential Myt1 kinase inhibitors by TR-FRET based binding assay. Eur. J. Med. Chem. 2013, 61, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Rohe, A.; Henze, C.; Erdmann, F.; Sippl, W.; Schmidt, M. A fluorescence anisotropy-based Myt1 kinase binding assay. Assay Drug Dev. Technol. 2014, 12, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, O.; Bomer, U. High-throughput screening for kinase inhibitors. Chembiochem 2005, 6, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Rohe, A.; Platzer, C.; Masch, A.; Greiner, S.; Henze, C.; Ihling, C.; Erdmann, F.; Schutkowski, M.; Sippl, W.; Schmidt, M. Identification of peptidic substrates for the human kinase Myt1 using peptide microarrays. Bioorg. Med. Chem. 2015, 23, 4936–4942. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Torrent, M.; Florjancic, A.S.; Bromberg, K.D.; Buchanan, F.G.; Ferguson, D.C.; Johnson, E.F.; Lasko, L.M.; Maag, D.; Merta, P.J.; et al. Pyrimidine-based tricyclic molecules as potent and orally efficacious inhibitors of wee1 kinase. ACS Med. Chem. Lett. 2015, 6, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Tibes, R.; Bogenberger, J.M.; Chaudhuri, L.; Hagelstrom, R.T.; Chow, D.; Buechel, M.E.; Gonzales, I.M.; Demuth, T.; Slack, J.; Mesa, R.A.; et al. RNAi screening of the kinome with cytarabine in leukemias. Blood 2012, 119, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.T.; Edwards, H.; Buck, S.A.; Ge, Y.; Taub, J.W. Targeting the wee1 kinase for treatment of pediatric down syndrome acute myeloid leukemia. Pediatr. Blood Cancer 2014, 61, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Yamanaka, K.; Itadani, H.; Arai, T.; Nishibata, T.; Hirai, H.; Kotani, H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol. Cancer 2009, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Cheng, R.S.; Mok, S.C. Identification of differentially expressed genes from ovarian cancer cells by MICROMAX cDNA microarray system. Biotechniques 2001, 30, 670–675. [Google Scholar] [PubMed]
- Seidl, C.; Port, M.; Gilbertz, K.P.; Morgenstern, A.; Bruchertseifer, F.; Schwaiger, M.; Roper, B.; Senekowitsch-Schmidtke, R.; Abend, M. 213Bi-induced death of HSC45-M2 gastric cancer cells is characterized by G2 arrest and up-regulation of genes known to prevent apoptosis but induce necrosis and mitotic catastrophe. Mol. Cancer Ther. 2007, 6, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. https://doi.org/10.3390/molecules22122045
Schmidt M, Rohe A, Platzer C, Najjar A, Erdmann F, Sippl W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules. 2017; 22(12):2045. https://doi.org/10.3390/molecules22122045
Chicago/Turabian StyleSchmidt, Matthias, Alexander Rohe, Charlott Platzer, Abdulkarim Najjar, Frank Erdmann, and Wolfgang Sippl. 2017. "Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases" Molecules 22, no. 12: 2045. https://doi.org/10.3390/molecules22122045
APA StyleSchmidt, M., Rohe, A., Platzer, C., Najjar, A., Erdmann, F., & Sippl, W. (2017). Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules, 22(12), 2045. https://doi.org/10.3390/molecules22122045