Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions




Abstract
:
1. Introduction
2. Codes for Annotation of Peptide Sequences and Structures
2.1. Annotation of Peptides Using Biological Codes
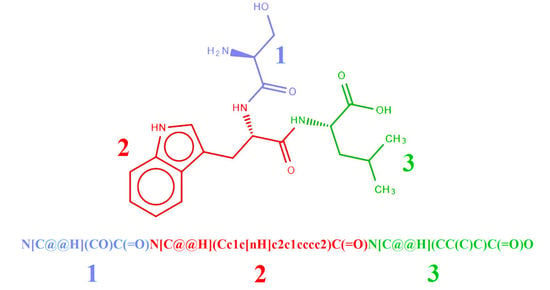
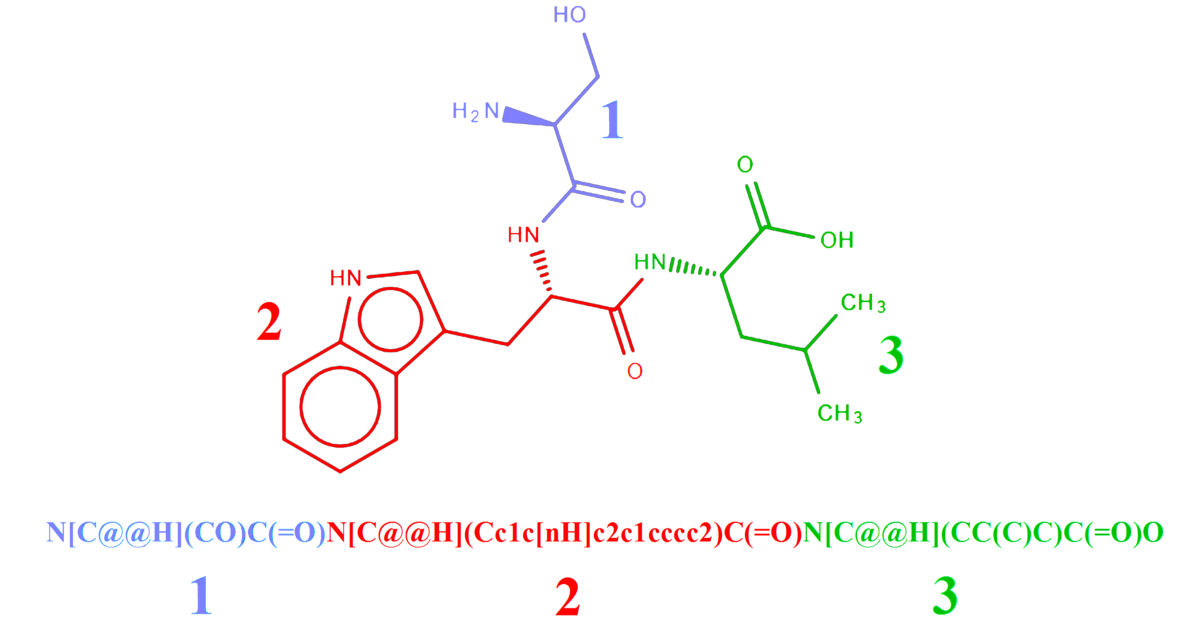
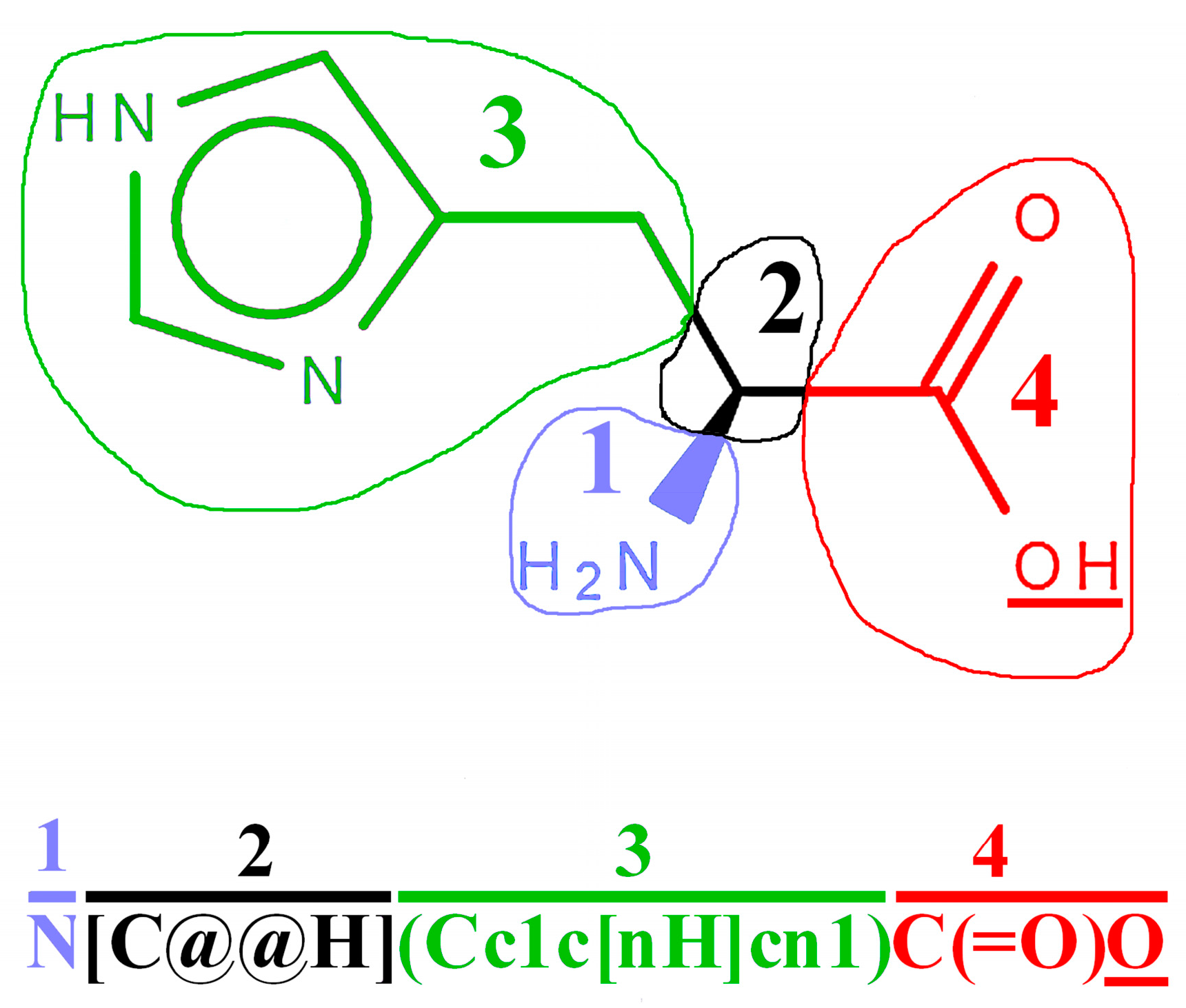
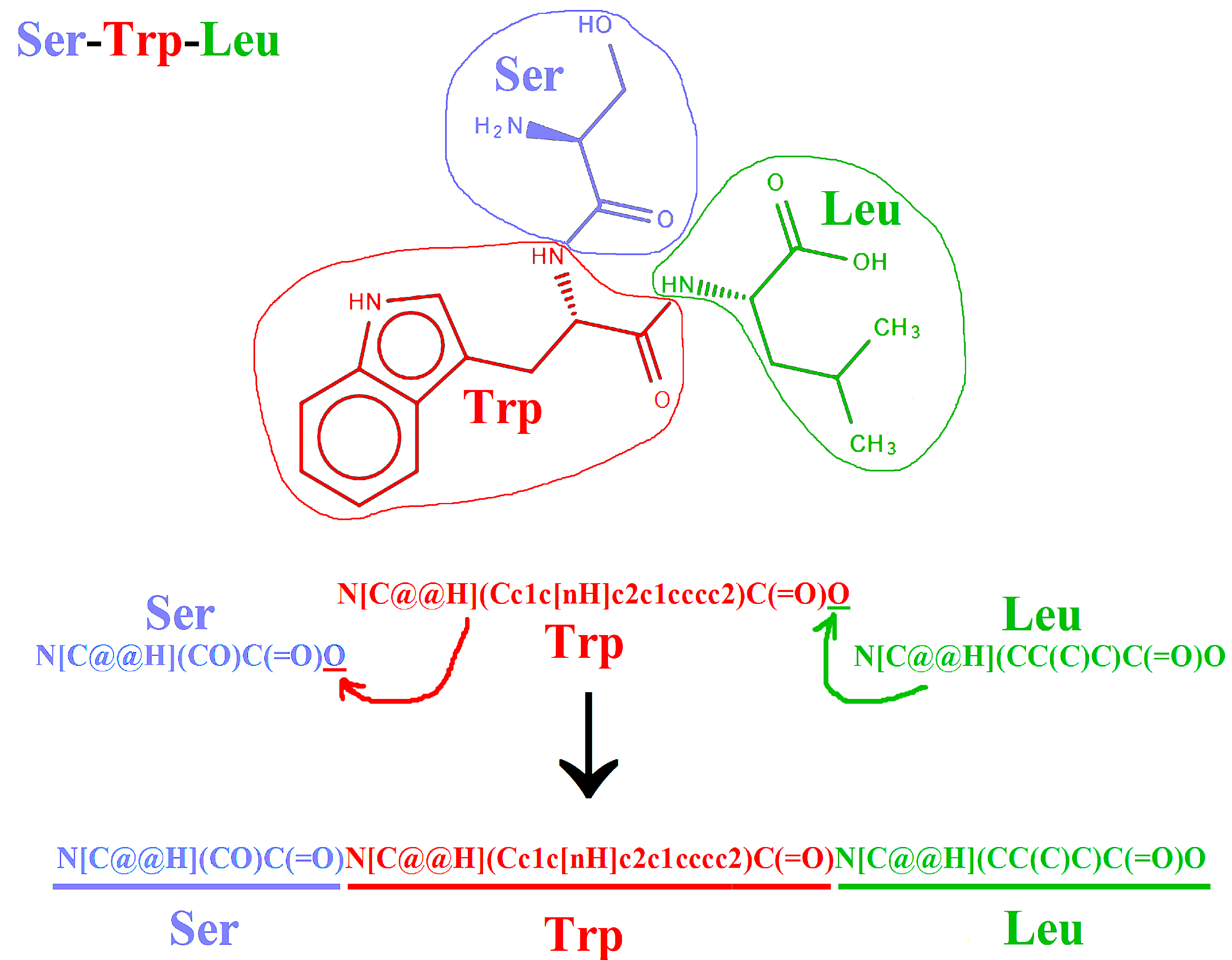
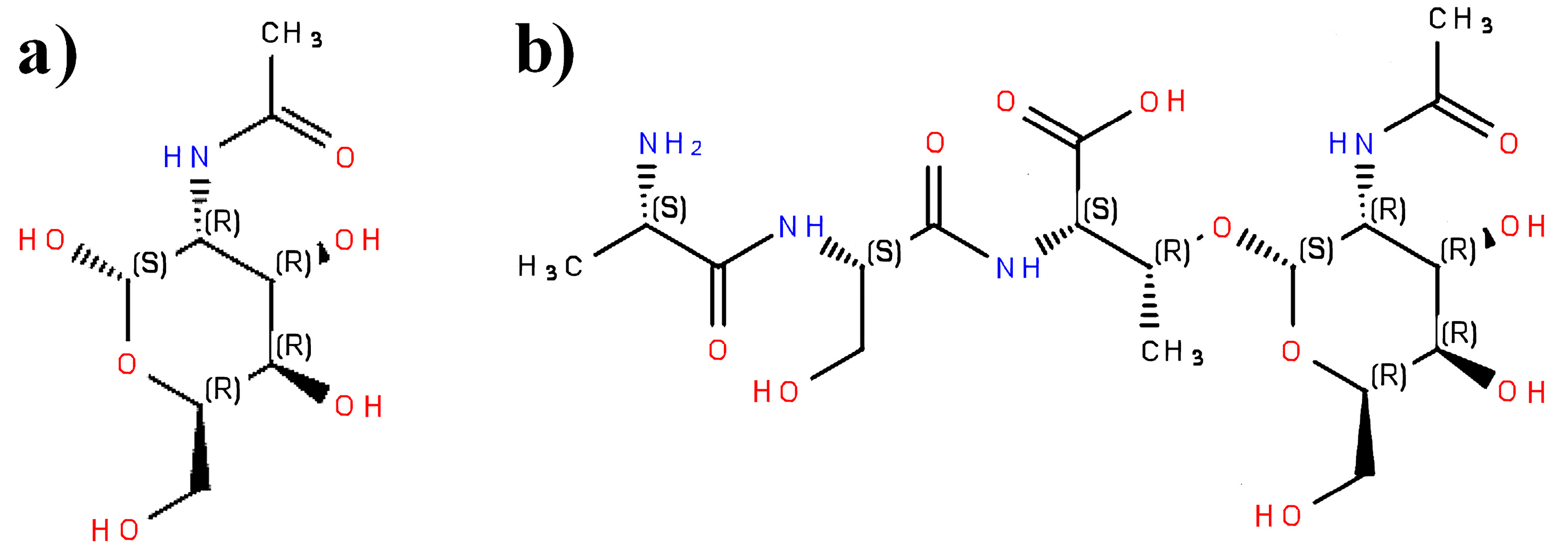
2.2. Representation of Peptides Using SMILES Code
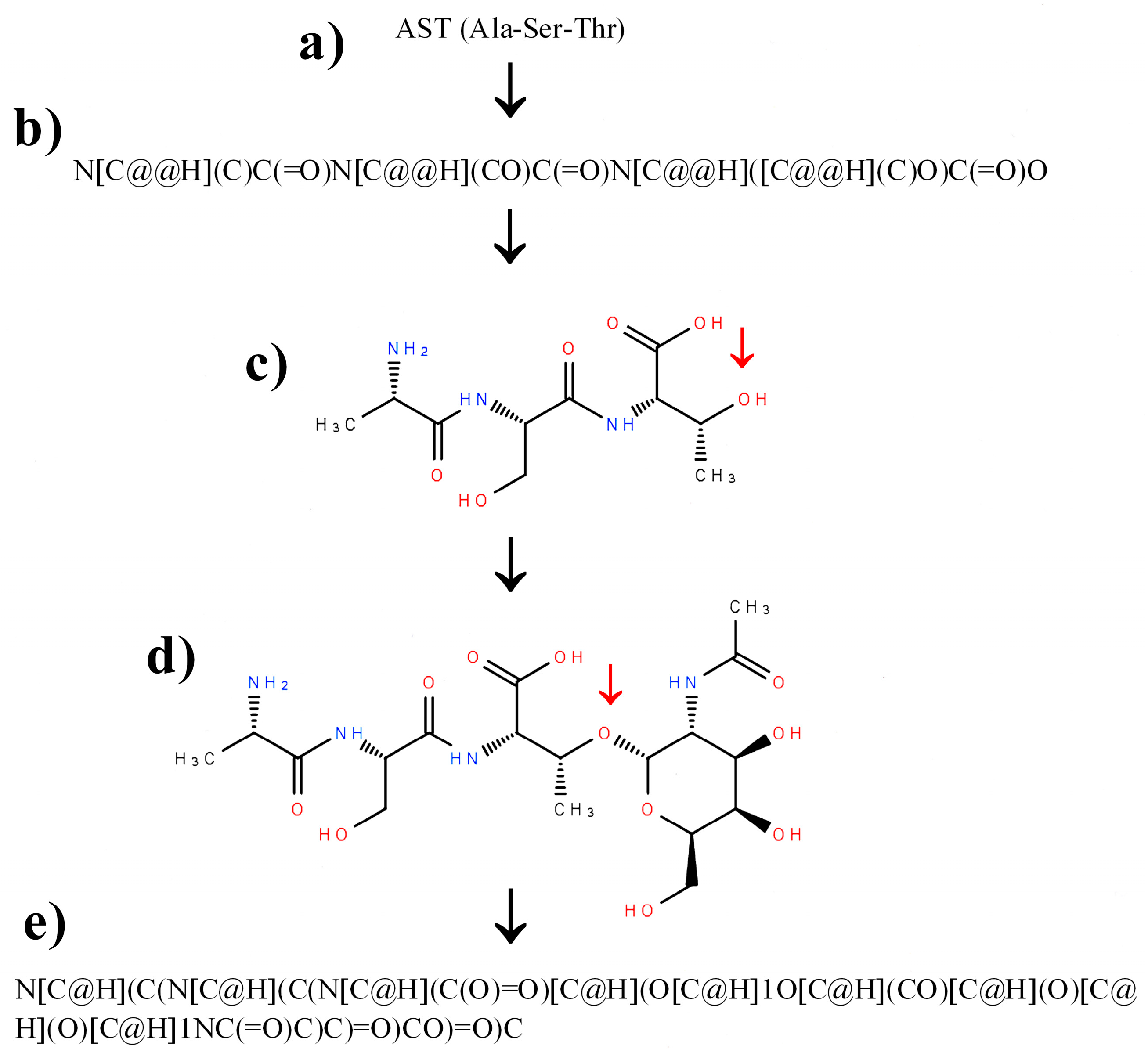
2.3. Construction of SMILES Representations of Peptide Containing Modified Amino Acid Residues Using Glycosylated Amino Acids as an Example
2.4. Other Chemical Codes
3. Verification and Correction of Errors in Peptide Structures Annotated Using SMILES Code
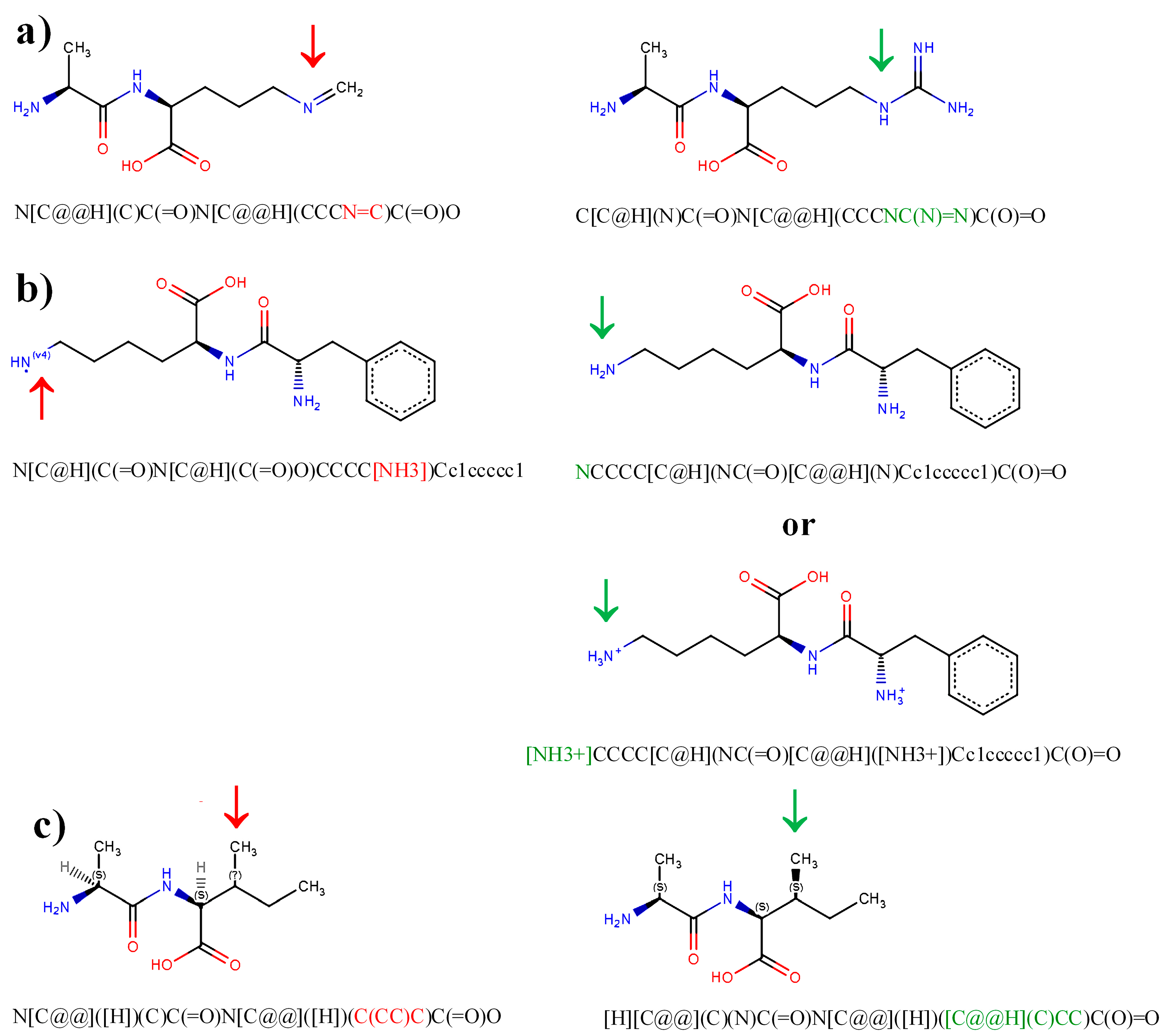
3.1. Typical Errors in Peptide Representations and Their Correction
3.2. Verification and Correction of Representations of Non-Peptidic Moiteties with Special Attention on Sugar Residues
4. Brief Recommendations for Training
5. Final Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADME | Absorption, Distribution, Metabolism and Excretion |
| AHTPDB | Antihypertensive peptide database |
| CID | Compound Identifier (in PubChem database) |
| EMBL | European Molecular Biology Laboratory |
| α-GalNAc | N-Acetyl-α-d-galactosamine–PubChem CID 84265 |
| HELM | Hierarchical Editing Language for Macromolecules |
| HMDB | Human Metabolome Database |
| InChI | International Chemical Identifier |
| InChIKey | Key of International Chemical Identifier |
| IUPAC | International Union of Pure and Applied Chemistry |
| LINUCS | Linear Notation for Unique Description of Carbohydrate Sequences |
| LIPID MAPS | Lipid Metabolites nd Pathways Strategy |
| MDL | Molecular Design Limited Inc. (company name) |
| QSAR | Quantitative Structure-Activity Relationship |
| R | One-letter symbol of amino acid arginine or absolute configuration of substituents around asymmetric carbon atom-Rectus (depending on context) |
| S | One-letter symbol of amino acid serine or absolute configuration of substituents around asymmetric carbon atom-Sinister (depending on context) |
| SATPdb | Structurally Annotated Therapeutic Peptide database |
| SMILES | Simplified Molecular Input Line Entry System or Simplified Molecular Input Line Entry Specification |
| WURCS | Web3 Unique Representation of Carbohydrate Structures |
References
- Senthilkumar, B.; Rajasekaran, R. Computational resources for designing peptide based drugs preferred in the field of nanomedicine. J. Bionanosci. 2016, 10, 1–14. [Google Scholar] [CrossRef]
- Siani, M.A.; Weininger, D.; Blaney, J.M. CHUCKLES: A method for representing and searching peptide and peptoid sequences on both monomer and atomic levels. J. Chem. Inf. Comput. Sci. 1994, 34, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, P.; Darewicz, M.; Iwaniak, A.; Bucholska, J.; Starowicz, P.; Czyrko, E. Internet databases of the properties, enzymatic reactions, and metabolism of small molecules-search options and applications in food science. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M.; Hrynkiewicz, M. Food protein-originating peptides as tastants—Physiological, technological, sensory, and bioinformatic approaches. Food Res. Int. 2016, 89, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- OpenSMILES. Available online: http://opensmiles.org/ (accessed on 29 September 2017).
- Ortiz-Martinez, M.; Gonzalez de Mejia, E.; García-Lara, S.; Aguilar, O.; Lopez-Castillo, L.M.; Otero-Pappatheodorou, J.T. Antiproliferative effect of peptide fractions isolated from a quality protein maize, a white hybrid maize, and their derived peptides on hepatocarcinoma human HepG2 cells. J. Funct. Foods 2017, 34, 36–48. [Google Scholar] [CrossRef]
- SwissTargetPrediction Program. Available online: http://www.swisstargetprediction.ch/ (accessed on 29 September 2017).
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Deming, T.J. Functional modification of thioether groups in peptides, polypeptides, and proteins. Bioconj. Chem. 2017, 28, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Obexer, R.; Walport, L.J.; Suga, H. Exploring sequence space: Harnessing chemical and biological diversity towards new peptide leads. Curr. Opin. Chem. Biol. 2017, 38, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Mehta, G. Late stage modification of peptides via CAH activation reactions. Tetrahedron Lett. 2017, 58, 1357–1372. [Google Scholar] [CrossRef]
- Stone, T.A.; Deber, C.M. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim. Biophys. Acta Biomembr. 2017, 1859, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Chingle, R.; Proulx, C.; Lubell, W.D. Azapeptide synthesis methods for expanding side-chain diversity for biomedical applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- PepstrMod. Available online: http://osddlinux.osdd.net/raghava/pepstrmod/ (accessed on 14 November 2017).
- Singh, S.; Singh, H.; Tuknait, A.; Chaudhary, K.; Singh, B.; Kumaran, S.; Raghava, G.P.S. PEPstrMOD: Structure prediction of peptides containing nat:ural, non-natural and modified residues. Biol. Direct 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.-L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floris, M.; Moro, S. Mimicking peptides… In silico. Mol. Inf. 2012, 31, 12–20. [Google Scholar] [CrossRef] [PubMed]
- BioTriangle Program. Available online: http://biotriangle.scbdd.com/ (accessed on 29 September 2017).
- Dong, J.; Yao, Z.-J.; Wen, M.; Zhu, M.-F.; Wang, N.-N.; Miao, H.-Y.; Lu, A.-P.; Zeng, W.-B.; Cao, D.-S. BioTriangle: A web-accessible platform for generating various molecular representations for chemicals, proteins, DNAs/RNAs and their interactions. J. Cheminform. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Swiss Institute of Bioinformatics. Available online: http://www.sib.swiss/services-resources/databases-tools (accessed on 29 September 2017).
- SwissADME Program. Available online: http://www.swissadme.ch/ (accessed on 29 September 2017).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- WebMolCS Program. Available online: http://www.gdbtools.unibe.ch:8080/webMolCS/ (accessed on 29 September 2017).
- Awale, M.; Probst, D.; Reymond, J.-L. WebMolCS: A web-based interface for visualizing molecules in three-dimensional chemical spaces. J. Chem. Inf. Model. 2017, 57, 643–649. [Google Scholar] [CrossRef] [PubMed]
- University of Bern. Available online: http://www.gdb.unibe.ch/ (accessed on 29 September 2017).
- ChemAxon. Available online: https://www.chemaxon.com/ (accessed on 14 November 2017).
- Brainpeps Database. Available online: http://brainpeps.ugent.be/ (accessed on 29 September 2017).
- Van Dorpe, S.; Bronselaer, A.; Nielandt, J.; Stalmans, S.; Wynendaele, E.; Audenaert, K.; Van De Wiele, C.; Burvenich, C.; Peremans, K.; Hsuchou, H.; et al. Brainpeps: The blood-brain barrier peptide database. Brain Struct. Funct. 2012, 217, 687–718. [Google Scholar] [CrossRef] [PubMed]
- Qorumpeps Database. Available online: http://quorumpeps.ugent.be/ (accessed on 9 November 2017).
- Wynendaele, E.; Bronselaer, A.; Nielandt, J.; D’Hondt, M.; Stalmans, S.; Bracke, N.; Verbeke, F.; Van De Wiele, C.; De Tré, G.; De Spiegeleer, B. Quorumpeps database: Chemical space, microbial origin and functionality of quorum sensing peptides. Nucleic Acids Res. 2013, 41, D655–D659. [Google Scholar] [CrossRef] [PubMed]
- AHTPDB Database. Available online: http://crdd.osdd.net/raghava/ahtpdb/ (accessed on 15 November 2017).
- Kumar, R.; Chaudhary, K.; Sharma, M.; Nagpal, G.; Chauhan, J.S.; Singh, S.; Gautam, A.; Raghava, G.P.S. AHTPDB: A comprehensive platform for analysis and presentation of antihypertensive peptides. Nucleic Acids Res. 2015, 43, D956–D962. [Google Scholar] [CrossRef] [PubMed]
- CancerPPD Database. Available online: http://crdd.osdd.net/raghava/cancerppd/index.php (accessed on 15 November 2017).
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P.S. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [PubMed]
- Hemolytik Database. Available online: http://crdd.osdd.net/raghava/hemolytik/ (accessed on 15 November 2017).
- Gautam, A.; Chaudhary, K.; Singh, S.; Joshi, A.; Anand, P.; Tuknait, A.; Mathur, D.; Varshney, G.C.; Raghava, G.P.S. Hemolytik: A database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. 2014, 42, D444–D449. [Google Scholar] [CrossRef] [PubMed]
- ParaPep Database. Available online: http://crdd.osdd.net/raghava/parapep/ (accessed on 15 November 2017).
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014. [Google Scholar] [CrossRef] [PubMed]
- PepLife Database. Available online: http://crdd.osdd.net/raghava/peplife/ (accessed on 15 November 2017).
- Mathur, D.; Prakash, S.; Anand, P.; Kaur, H.; Agrawal, P.; Mehta, A.; Kumar, R.; Singh, S.; Raghava, G.P.S. PEPlife: A repository of the halflife of peptides. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- SATPdb Database. Available online: http://crdd.osdd.net/raghava/satpdb/links.php (accessed on 29 September 2017).
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P.S. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016, 44, D1119–D1126. [Google Scholar] [CrossRef] [PubMed]
- BIOPEP Database. Available online: http://www.uwm.edu.pl/biochemia/index.php/pl/biopep (accessed on 29 September 2017).
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M.; Sieniawski, K.; Starowicz, P. BIOPEP database of sensory peptides and amino acids. Food Res. Int. 2016, 85, 155–161. [Google Scholar] [CrossRef]
- OmicTools. Available online: https://omictools.com/ (accessed on 29 September 2017).
- Henry, V.J.; Bandrowski, A.E.; Pepin, A.-S.; Gonzalez, B.J.; Desfeux, A. OMICtools: An informative directory for multi-omic data analysis. Database 2014. [Google Scholar] [CrossRef] [PubMed]
- MetaComBio. Available online: http://www.uwm.edu.pl/metachemibio/index.php/about-metacombio (accessed on 29 September 2017).
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. Using internet databases for food science organic chemistry students to discover chemical compound information. J. Chem. Educ. 2015, 92, 874–876. [Google Scholar] [CrossRef]
- LabWorm. Available online: https://labworm.com/ (accessed on 29 September 2017).
- Udenigwe, C.C. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci. Technol. 2014, 36, 137–143. [Google Scholar] [CrossRef]
- PEP-FOLD Program. Available online: http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/ (accessed on 10 November 2017).
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- (PS)2 v3. Available online: http://ps2v3.life.nctu.edu.tw/ (accessed on 10 November 2017).
- Huang, T.-T.; Hwang, J.-K.; Chen, C.-H.; Chu, C.-S.; Lee, C.-W.; Chen, C.-C. (PS)2: Protein structure prediction server version 3.0. Nucleic Acids Res. 2015, 43, W338–W342. [Google Scholar] [CrossRef] [PubMed]
- Iwaniak, A.; Minkiewicz, P.; Darewicz, M.; Protasiewicz, M.; Mogut, D. Chemometrics and cheminformatics in the analysis of biologically active peptides from food sources. J. Funct. Foods 2015, 16, 334–351. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; FitzGerald, R.J. Learnings from quantitative structure-activity relationship (QSAR) studies with respect to food protein-derived bioactive peptides: A review. RSC Adv. 2016, 6, 75400–75413. [Google Scholar] [CrossRef]
- Chou, K.-C. Prediction of protein cellular attributes using pseudo-amino-acid-composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C. Pseudo amino acid composition and its application in bioinformatics, proteomics and system biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. Recent progress in protein subcellular localization. Anal. Biochem. 2007, 370, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Ekins, S. A quality alert and call for improved curation of public chemistry databases. Drug Discov. Today 2011, 16, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- SwissSidechain Database. Available online: http://swisssidechain.ch/ (accessed on 29 September 2017).
- Gfeller, D.; Michielin, O.; Zoete, V. SwissSidechain: A molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013, 41, D327–D332. [Google Scholar] [CrossRef] [PubMed]
- Norine Database. Available online: http://bioinfo.lifl.fr/NRP/ (accessed on 29 September 2017).
- Flissi, A.; Dufresne, Y.; Michalik, J.; Tonon, L.; Janot, S.; Noé, L.; Jacques, P.; Leclère, V.; Pupin, M. Norine, the knowledgebase dedicated to non-ribosomal peptides, is now open to crowdsourcing. Nucleic Acids Res. 2016, 44, D1113–D1118. [Google Scholar] [CrossRef] [PubMed]
- CycloPS Program. Available online: http://bioware.ucd.ie/~cyclops/cgi-bin/webpep.cgi (accessed on 29 September 2017).
- Duffy, F.J.; Verniere, M.; Devocelle, M.; Bernard, E.; Shields, D.C.; Chubb, A.J. CycloPs: Generating virtual libraries of cyclized and constrained peptides including nonnatural amino acids. J. Chem. Inf. Model. 2011, 51, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Bohne-Lang, A.; Lang, E.; Förster, T.; von der Lieth, C.-W. LINUCS: Linear notation for unique description of carbohydrate sequences. Carbohydr. Res. 2000, 336, 1–11. [Google Scholar] [CrossRef]
- PubChem Database. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 29 September 2017).
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, H.; Xi, H.; Stanton, R.V.; Rotstein, S.H. HELM: A hierarchical notation language for complex biomolecule structure representation. J. Chem. Inf. Model. 2012, 52, 2796–2806. [Google Scholar] [CrossRef] [PubMed]
- Milton, J.; Zhang, T.; Bellamy, C.; Swayze, E.; Hart, C.; Weisser, M.; Hecht, S.; Rotstein, S. HELM software for biopolymers. J. Chem. Inf. Model. 2017, 57, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL Database. Available online: https://www.ebi.ac.uk/chembldb/ (accessed on 29 September 2017).
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, M.; Jin, X.; Marcaida, M.J.; Banha, J.; Dibonaventura, I.; Bongoni, S.; Bartho, K.; Gräbner, O.; Sefkow, M.; Darbre, T.; et al. Bridged bicyclic peptides as potential drug scaffolds: Synthesis, structure, protein binding and stability. Chem. Sci. 2015, 6, 5473–5490. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- ChemSpider Database. Available online: http://www.chemspider.com/Default.aspx (accessed on 29 September 2017).
- Williams, A.; Tkachenko, V. The royal society of chemistry and the delivery of chemistry data repositories for the community. J. Comput. Aided Mol. Des. 2014, 28, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- ZINC 15 Database. Available online: http://zinc15.docking.org/ (accessed on 29 September 2017).
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- HMDB Database. Available online: http://www.hmdb.ca/ (accessed on 29 September 2017).
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Ketcher Program. Available online: http://lifescience.opensource.epam.com/ketcher/ (accessed on 29 September 2017).
- Karulin, B.; Kozhevnikov, M. Ketcher: Web-based chemical structure editor. J. Cheminform. 2011, 3 (Suppl. S1). [Google Scholar] [CrossRef]
- Open Babel Program. Available online: http://openbabel.org/wiki/Main_Page (accessed on 29 September 2017).
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 2000, 132, 185–219. [Google Scholar] [PubMed]
- Wikipedia FASTA Format. Available online: https://en.wikipedia.org/wiki/FASTA_format (accessed on 29 September 2017).
- UniProt Database. Available online: http://www.uniprot.org/ (accessed on 29 September 2017).
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Wkipedia Protein Data Bank File Format. Available online: https://en.wikipedia.org/wiki/Protein_Data_Bank_(file_format) (accessed on 9 November 2017).
- Ertl, P. Molecular structure input on the web. J. Cheminform. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Okerblom, J.; Varki, A. Biochemical, cellular, physiological, and pathological consequences of human loss of N-glycolylneuraminic acid. ChemBioChem 2017, 18, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Palaniappan, K.K.; Bertozzi, C.R. Chemical glycoproteomics. Chem. Rev. 2016, 116, 14277–14306. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M. Basic primitives for molecular diagram sketching. J. Cheminform. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. Specification of molecular chirality. Angew. Chem. Int. Ed. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Wikipedia Cahn–Ingold–Prelog Priority Rules. Available online: https://en.wikipedia.org/wiki/Cahn%E2%80%93Ingold%E2%80%93Prelog_priority_rules (accessed on 29 September 2017).
- Chemical Identifier Resolver Program. Available online: https://cactus.nci.nih.gov/chemical/structure (accessed on 29 September 2017).
- Muresan, S.; Sitzmann, M.; Southan, C. Mapping between databases of compounds and protein targets. Meth. Mol. Biol. 2012, 910, 145–164. [Google Scholar] [CrossRef]
- Chemical Translation Service Program. Available online: http://cts.fiehnlab.ucdavis.edu/ (accessed on 29 September 2017).
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The chemical translation service—A web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef] [PubMed]
- Smiles2Monomers Program. Available online: http://bioinfo.lifl.fr/norine/smiles2monomers.jsp (accessed on 29 September 2017).
- Dufresne, Y.; Noé, L.; Leclère, V.; Pupin, M. Smiles2Monomers: A link between chemical and biological structures for polymers. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Southan, C. InChI in the wild: An assessment of InChIKey searching in Google. J. Cheminform. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Warr, W.A. Many InChIs and quite some feat. J. Comput. Aided Mol. Des. 2015, 29, 681–694. [Google Scholar] [CrossRef] [PubMed]
- SwissDock Program. Available online: http://www.swissdock.ch/ (accessed on 29 September 2017).
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- AMMOS2 Program. Available online: http://drugmod.rpbs.univ-paris-diderot.fr/ammosHome.php (accessed on 29 September 2017).
- Labbé, C.; Pencheva, T.; Jereva, D.; Desvillechabrol, D.; Becot, J.; Villoutreix, B.O.; Pajeva, I.; Miteva, M.A. AMMOS2: A web server for protein-ligand-water complexes refinement via molecular mechanics. Nucleic Acids Res. 2017, 45, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- ProteinsPlus Program. Available online: http://proteinsplus.zbh.uni-hamburg.de/ (accessed on 29 September 2017).
- Fährrolfes, R.; Bietz, S.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Otto, T.; Volkamer, A.; Rarey, M. ProteinsPlus: A web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017, 45, W337–W343. [Google Scholar] [CrossRef] [PubMed]
- LIPID MAPS® Database. Available online: http://www.lipidmaps.org/ (accessed on 29 September 2017).
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.P.; Ranzinger, R.; Lütteke, T.; Mariethoz, J.; Hayes, C.A.; Zhang, J.; Akune, Y.; Aoki-Kinoshita, K.F.; Damerell, D.; Carta, G.; et al. Toolboxes for a standardised and systematic study of glycans. BMC Bioinform. 2014, 15 (Suppl. S1). [Google Scholar] [CrossRef] [PubMed]
- WURCS Program. Available online: http://www.wurcs-wg.org/software.php (accessed on 29 September 2017).
- Matsubara, M.; Aoki-Kinoshita, K.F.; Aoki, N.P.; Yamada, I.; Narimatsu, H. WURCS 2.0 update to encapsulate ambiguous carbohydrate structures. J. Chem. Inf. Model. 2017, 57, 632–637. [Google Scholar] [CrossRef] [PubMed]
- GlyTouCan Database. Available online: https://glytoucan.org/ (accessed on 29 September 2017).
- Aoki-Kinoshita, K.; Agravat, S.; Aoki, N.P.; Arpinar, S.; Cummings, R.D.; Fujita, A.; Fujita, N.; Hart, G.M.; Haslam, S.M.; Kawasaki, T.; et al. GlyTouCan 1.0—The international glycan structure repository. Nucleic Acids Res. 2016, 44, D1237–D1242. [Google Scholar] [CrossRef] [PubMed]
- Wikipedia Chemical Table File. Available online: https://en.wikipedia.org/wiki/Chemical_table_file#Molfile (accessed on 29 September 2017).
- Tanaka, K.; Aoki-Kinoshita, K.F.; Kotera, M.; Sawaki, H.; Tsuchiya, S.; Fujita, N.; Shikanai, T.; Kato, M.; Kawano, S.; Yamada, I.; et al. WURCS: The Web3 unique representation of carbohydrate structures. J. Chem. Inf. Model. 2014, 54, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- JCGGBD Database. Available online: http://jcggdb.jp/database_en.html (accessed on 29 September 2017).
- Maeda, M.; Fujita, N.; Suzuki, Y.; Sawaki, H.; Shikanai, T.; Narimatsu, H. JCGGDB: Japan consortium for glycobiology and glycotechnology database. Methods Mol. Biol. 2015, 1273, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Muratov, E.N.; Capuzzi, S.J.; Politi, R.; Low, Y.; Braga, R.C.; Zakharov, A.V.; Sedykh, A.; Mokshyna, E.; Farag, S.; et al. Alarms about structural alerts. Green Chem. 2016, 18, 4348–4360. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Engkvist, O.; Koch, U.; Reymond, J.-L.; Chen, H. BIGCHEM: Challenges and opportunities for big data analysis in chemistry. Mol. Inf. 2016, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Error | Consequences |
|---|---|
| Missed nitrogen atoms in guanidine groups | Possible errors in results of modeling interactions with biomacromolecules, inappropriate InChIKey, insufficient to be a query in database searching |
| Inappropriate valence of nitrogen atoms, e.g., in amine or guanidine groups | Possible errors in results of modeling interactions with biomacromolecules, unavailable for database search engines (e.g., for search engine of ZINC15 database), inappropriate InChIKey, insufficient to be a query in database searching |
| Undefined or inappropriate configuration of substituents around asymmetric carbon atom, e.g., C3 atom in isoleucine or threonine | Possible errors in results of modeling interactions with biomacromolecules, inappropriate InChIKey, insufficient to be a query in database searching |
| Spaces in SMILES strings | Disabled processing of SMILES strings by database search engines and other programs |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minkiewicz, P.; Iwaniak, A.; Darewicz, M. Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions. Molecules 2017, 22, 2075. https://doi.org/10.3390/molecules22122075
Minkiewicz P, Iwaniak A, Darewicz M. Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions. Molecules. 2017; 22(12):2075. https://doi.org/10.3390/molecules22122075
Chicago/Turabian StyleMinkiewicz, Piotr, Anna Iwaniak, and Małgorzata Darewicz. 2017. "Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions" Molecules 22, no. 12: 2075. https://doi.org/10.3390/molecules22122075
APA StyleMinkiewicz, P., Iwaniak, A., & Darewicz, M. (2017). Annotation of Peptide Structures Using SMILES and Other Chemical Codes–Practical Solutions. Molecules, 22(12), 2075. https://doi.org/10.3390/molecules22122075