The Design and Evaluation of an l-Dopa–Lazabemide Prodrug for the Treatment of Parkinson’s Disease


Abstract
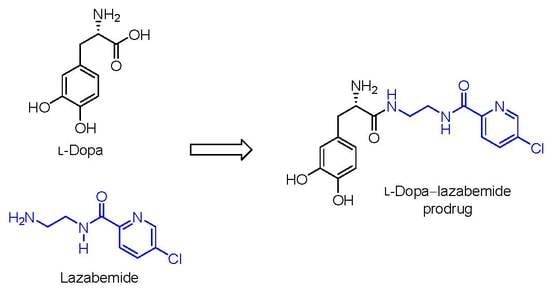
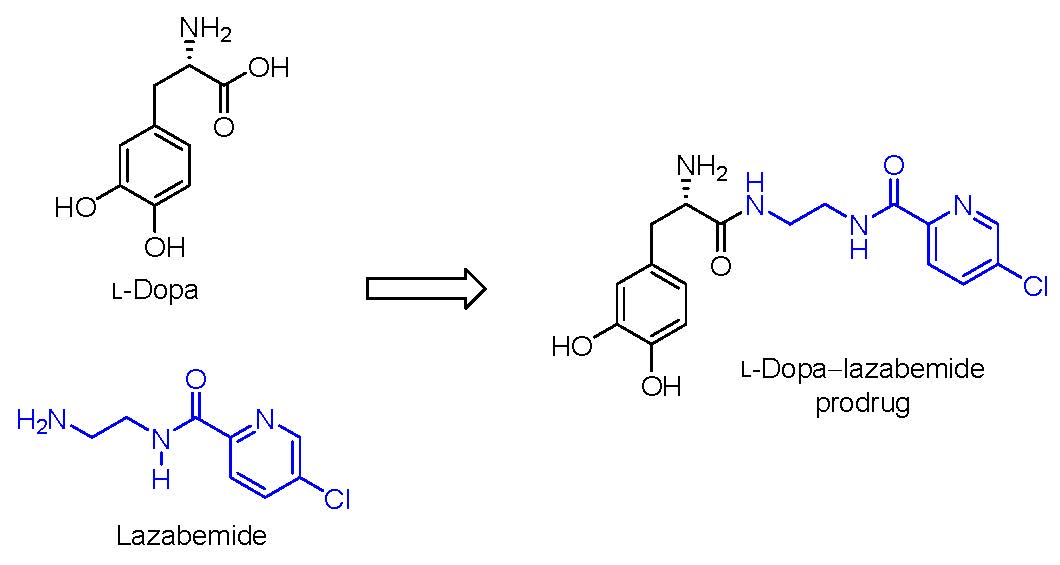
:
1. Introduction
2. Results
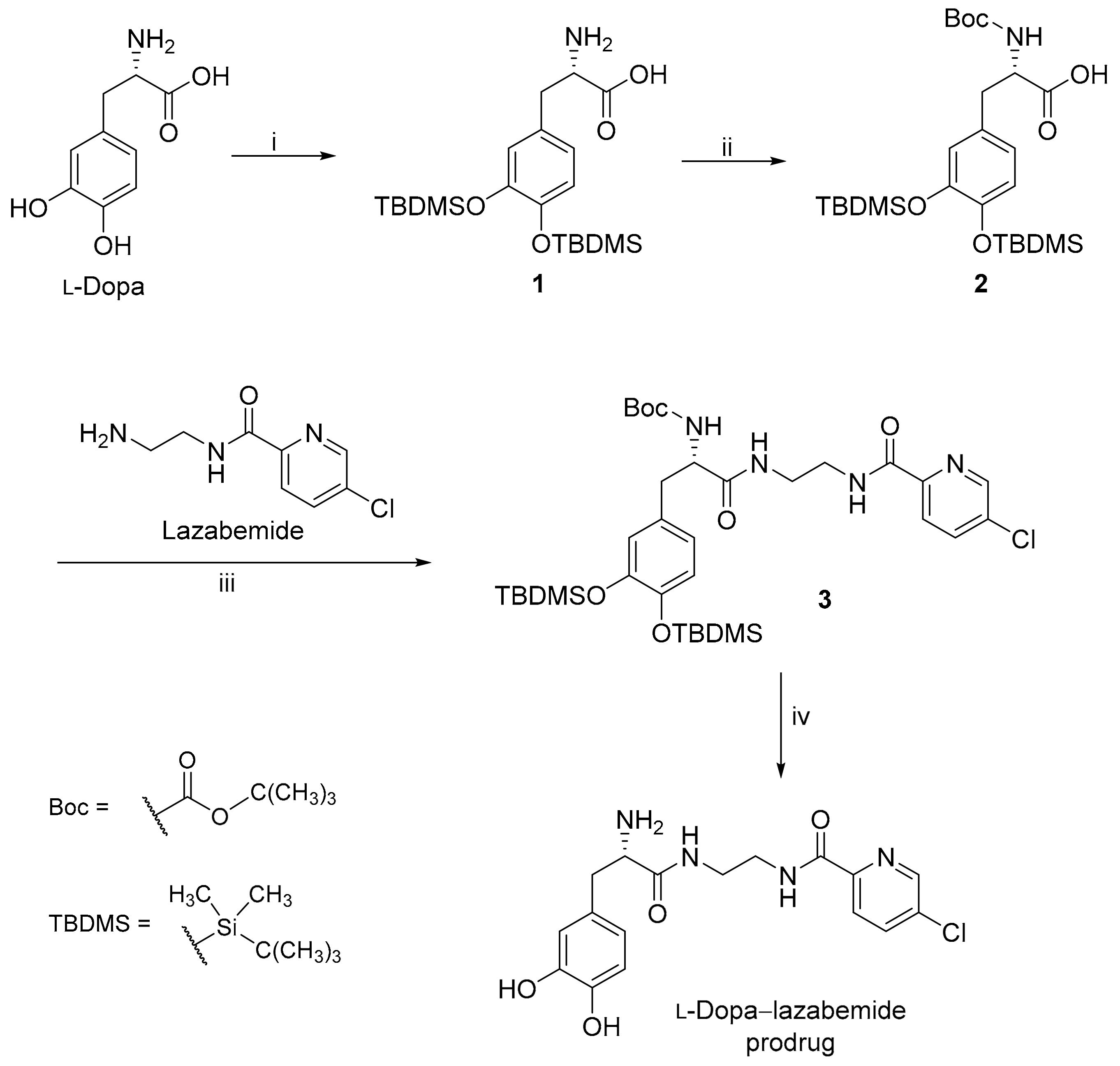
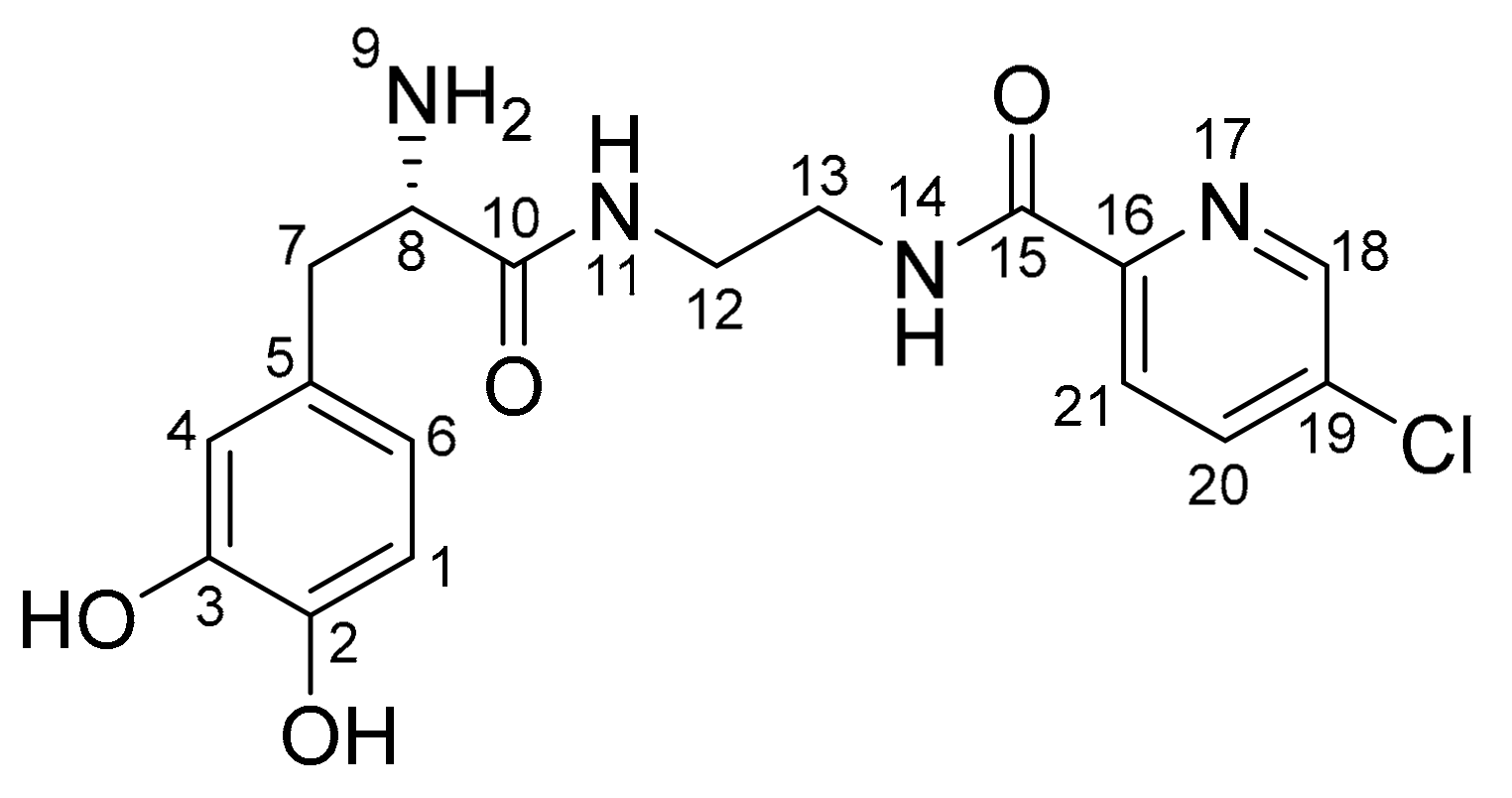
2.1. Synthesis of the l-Dopa–Lazabemide Prodrug
2.2. Physicochemical Properties
2.2.1. LogD
2.2.2. Solubility
2.2.3. Ionisation Constant
2.2.4. Cell Viability
2.2.5. Passive Diffusion Permeability
2.2.6. Chemical Stability
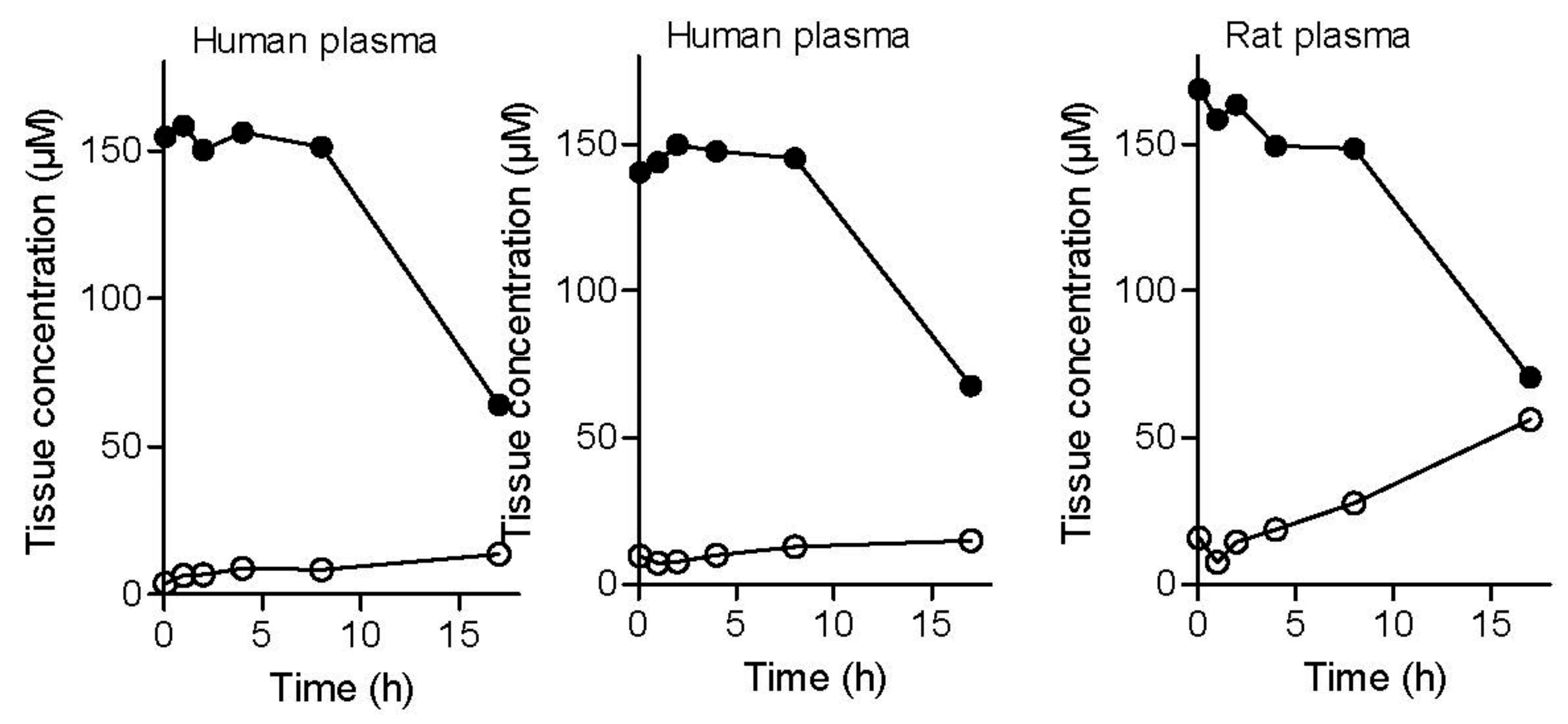
2.2.7. Plasma and Tissue Stability
2.3. In-Vivo Effect of the Prodrug on Brain Monoamines
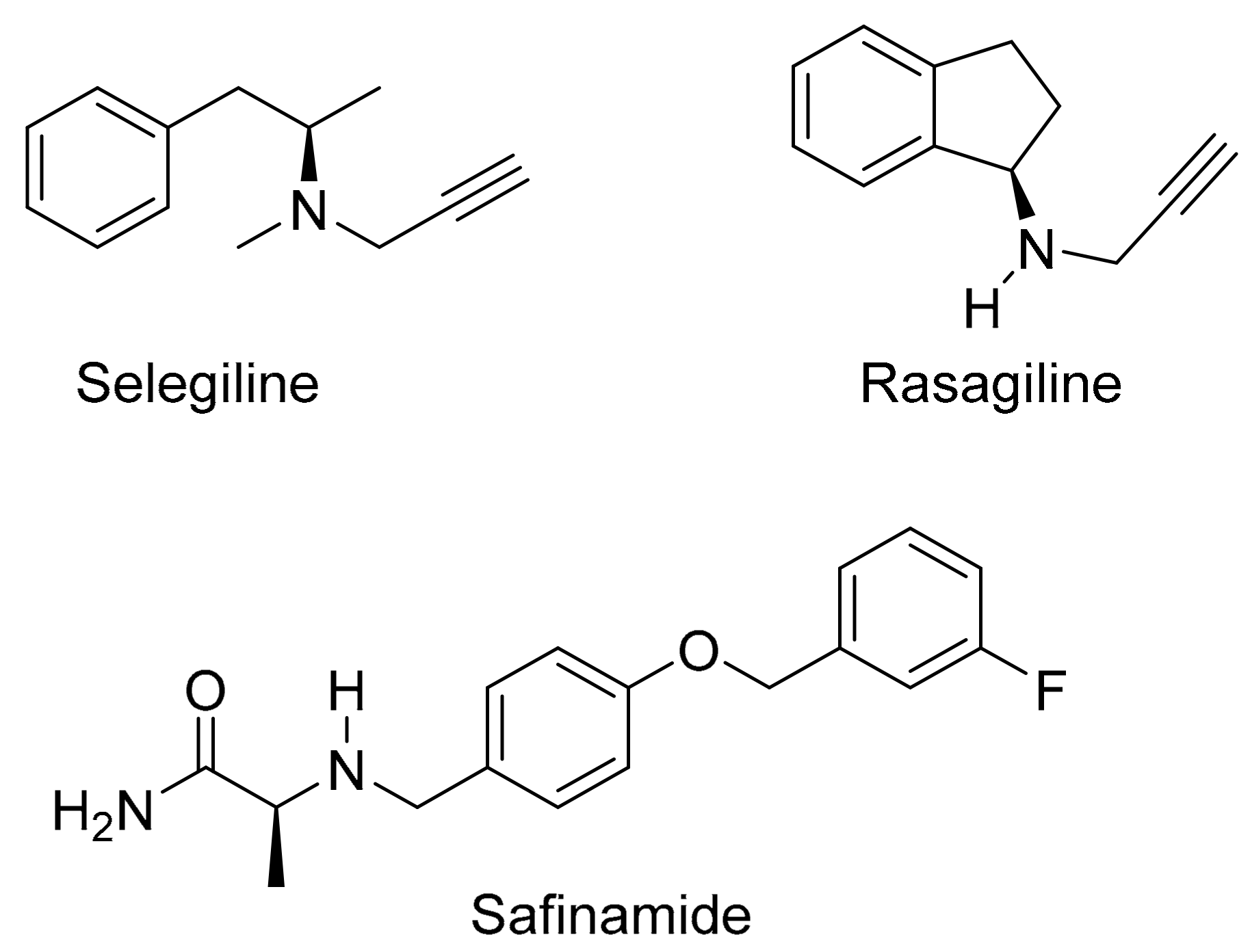
3. Discussion
4. Materials and Methods
4.1. The Synthesis of Lazabemide
4.2. The Synthesis of the l-Dopa–Lazabemide Prodrug
4.2.1. Chemicals and Instrumentation
4.2.2. The Synthesis of l-Dopa(TBDMS)2
4.2.3. The Synthesis of Boc-l-Dopa(TBDMS)2
4.2.4. The Conjugation of Protected l-Dopa with Lazabemide
4.2.5. Removal of the TBDMS and Boc Protective Groups from Boc-l-Dopa(TBDMS)2-lazabemide
4.3. Determination of Physicochemical and Biological Properties
4.3.1. Materials and Instrumentation
4.3.2. Ethics Consideration
4.3.3. Shake-Flask Method for logD Determination
4.3.4. Determination of Solubility
4.3.5. Determination of Ionisation Constant, pKa
4.3.6. Determination of Toxicity towards Cultured Cells
4.3.7. High-Performance Liquid Chromatography (HPLC)
4.3.8. Determination of Passive Diffusion Permeability
4.3.9. Determination of Chemical Stability
4.3.10. Determination of Plasma and Tissue Stability
4.4. Animal Studies
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Freitas, M.E.; Ruiz-Lopez, M.; Fox, S.H. Novel levodopa formulations for Parkinson’s disease. CNS Drugs 2016, 30, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Antonini, A. Novel formulations and modes of delivery of levodopa. Mov. Disord. 2015, 30, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Sozio, P.; Cerasa, L.S.; Iannitelli, A. l-Dopa prodrugs: An overview of trends for improving Parkinson’s disease treatment. Curr. Pharm. Des. 2011, 17, 3482–3493. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Sozio, P.; Cerasa, L.S. Antiparkinson prodrugs. Molecules 2008, 13, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Martinelli, P. Pharmacokinetics of levodopa. J. Neurol. 2010, 257, S253–S261. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, L.C.; Hauser, R.A. Carbidopa levodopa enteral suspension. Expert Opin. Pharmacother. 2015, 16, 2807–2817. [Google Scholar] [CrossRef] [PubMed]
- Nutt, J.G.; Fellman, J.H. Pharmacokinetics of levodopa. Clin. Neuropharmacol. 1984, 7, 35–49. [Google Scholar] [PubMed]
- Tohgi, H.; Abe, T.; Kikuchi, T.; Takahashi, S.; Nozaki, Y. The significance of 3-O-methyldopa concentrations in the cerebrospinal fluid in the pathogenesis of wearing-off phenomenon in Parkinson’s disease. Neurosci. Lett. 1991, 132, 19–22. [Google Scholar] [CrossRef]
- Learmonth, D.A.; Palma, P.N.; Vieira-Coelho, M.A.; Soares-da-Silva, P. Synthesis, biological evaluation, and molecular modeling studies of a novel, peripherally selective inhibitor of catechol-O-methyltransferase. J. Med. Chem. 2004, 47, 6207–6217. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, E.; Linden, I.B.; Schultz, E.; Pohto, P. Biochemical and pharmacological properties of a peripherally acting catechol-O-methyltransferase inhibitor entacapone. Naunyn Schmiedebergs Arch. Pharmacol. 1992, 346, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Shoulson, I.; Glaubiger, G.A.; Chase, T.N. On-off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology 1975, 25, 1144–1148. [Google Scholar] [PubMed]
- Nutt, J.G. On-off phenomenon: Relation to levodopa pharmacokinetics and pharmacodynamics. Ann. Neurol. 1987, 22, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Leenders, K.L.; Poewe, W.H.; Palmer, A.J.; Brenton, D.P.; Frackowiak, R.S. Inhibition of l-[18f]fluorodopa uptake into human brain by amino acids demonstrated by positron emission tomography. Ann. Neurol. 1986, 20, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Nutt, J.G.; Woodward, W.R.; Hammerstad, J.P.; Carter, J.H.; Anderson, J.L. The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N. Engl. J. Med. 1984, 310, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Lewitt, P.A.; Ellenbogen, A.; Chen, D.; Lal, R.; McGuire, K.; Zomorodi, K.; Luo, W.; Huff, F.J. Actively transported levodopa prodrug xp21279: A study in patients with Parkinson disease who experience motor fluctuations. Clin. Neuropharmacol. 2012, 35, 103–110. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Huff, F.J.; Hauser, R.A.; Chen, D.; Lissin, D.; Zomorodi, K.; Cundy, K.C. Double-blind study of the actively transported levodopa prodrug xp21279 in Parkinson’s disease. Mov. Disord. 2014, 29, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Cesura, A.M.; Muggli-Maniglio, D.; Lang, G.; Imhof, R.; Da Prada, M. Monoamine oxidase inhibition by moclobemide and 2-amino-ethyl carboxamide derivatives: Mode of action and kinetic characteristics. J. Neural Transm. Suppl. 1990, 32, 165–170. [Google Scholar] [PubMed]
- Cesura, A.M.; Borroni, E.; Gottowik, J.; Kuhn, C.; Malherbe, P.; Martin, J.; Richards, J.G. Lazabemide for the treatment of Alzheimer’s disease: Rationale and therapeutic perspectives. Adv. Neurol. 1999, 80, 521–528. [Google Scholar] [PubMed]
- Binda, C.; Li, M.; Hubálek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.E.; Mattevi, A.; Binda, C.; Li, M.; Hubálek, F. Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 2004, 11, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Dingemanse, J.; Wood, N.; Jorga, K.; Kettler, R. Pharmacokinetics and pharmacodynamics of single and multiple doses of the MAO-B inhibitor lazabemide in healthy subjects. Br. J. Clin. Pharmacol. 1997, 43, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Logan, J.; Schlyer, D.J.; MacGregor, R.R.; Wang, G.J.; Wolf, A.P.; Pappas, N.; Alexoff, D.; Shea, C. Monoamine oxidase B (MAO B) inhibitor therapy in Parkinson’s disease: The degree and reversibility of human brain MAO B inhibition by Ro 19 6327. Neurology 1993, 43, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.; Wang, J.; Bankiewicz, K.; Harvey-White, J.; Kopin, I.J.; Goldstein, D.S. Increased striatal dopamine production from l-DOPA following selective inhibition of monoamine oxidase B by R(+)-N-propargyl-1-aminoindan (rasagiline) in the monkey. J. Neural Transm. Suppl. 1998, 52, 279–285. [Google Scholar] [PubMed]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S287–S296. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Muller, T. Emerging approaches in Parkinson’s disease—Adjunctive role of safinamide. Ther. Clin. Risk Manag. 2016, 12, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Robakis, D.; Fahn, S. Defining the role of the monoamine oxidase-B inhibitors for Parkinson’s disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Olmstead, E.G.; Jacob, R.F. Antioxidant activity of the monoamine oxidase B inhibitor lazabemide. Biochem. Pharmacol. 2000, 60, 709–716. [Google Scholar] [CrossRef]
- Silverman, R.B. The Organic Chemistry of Drug Design and Drug Action, 2nd ed.; Elsevier Academic Press: Cambridge, MA, USA, 2004; p. 503. [Google Scholar]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization; Elsevier Academic Press: Cambridge, MA, USA, 2008; pp. 43–55. [Google Scholar]
- Berlin, I.; Aubin, H.J.; Pedarriosse, A.M.; Rames, A.; Lancrenon, S.; Lagrue, G. Lazabemide in Smoking Cessation Study Investigators. Lazabemide, a selective, reversible monoamine oxidase B inhibitor, as an aid to smoking cessation. Addiction 2002, 97, 1347–1354. [Google Scholar] [PubMed]
- Soriato, G.; Focati, M.P.; Brescello, R.; Cotarca, L.; Giovanetti, R. Pharmaceutical Preparations of Crystalline Lazabemide. Patent WO/2008/010794, 24 January 2008. [Google Scholar]
- Nakonieczna, L.; Przychodzeń, W.; Chimiak, A. A new convenient route for the synthesis of dopa peptides. Liebigs Ann. Chem. 1994, 1994, 1055–1058. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Brink, C.B.; Pretorius, A.; van Niekerk, B.P.; Oliver, D.W.; Venter, D.P. Studies on cellular resilience and adaptation following acute and repetitive exposure to ozone in cultured human epithelial (HeLa) cells. Redox Rep. 2008, 13, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Wohnsland, F.; Faller, B. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 2001, 44, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Hider, R.C.; Jenner, P.; Campbell, B.; Hobbs, C.J.; Rose, S.; Jairaj, M.; Tayarani-Binazir, K.A.; Syme, A. Design, synthesis and biological evaluation of l-dopa amide derivatives as potential prodrugs for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2010, 45, 4035–4042. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.H.; Brand, L.; Jeeva, Z.; Stein, D.J. Cortical/hippocampal monoamines, HPA-axis changes and aversive behavior following stress and restress in an animal model of post-traumatic stress disorder. Physiol. Behav. 2006, 87, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Kaenmaki, M.; Tammimaki, A.; Garcia-Horsman, J.A.; Myohanen, T.; Schendzielorz, N.; Karayiorgou, M.; Gogos, J.A.; Mannisto, P.T. Importance of membrane-bound catechol-O-methyltransferase in l-dopa metabolism: A pharmacokinetic study in two types of COMT gene modified mice. Br. J. Pharmacol. 2009, 158, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.; Serjeant, E.P. The Determination of Ionization Constants. A Laboratory Manual, 3rd ed.; Chapman and Hall: New York, NY, USA, 1984; pp. 14–38. ISBN 978-94-010-8948-7. [Google Scholar]
- Barrow, P.C.; Holt, S.J. Differences in distribution of esterase between cell fractions of rat liver homogenates prepared in various media. Relevance to the lysosomal location of the enzyme in the intact cell. Biochem. J. 1971, 125, 545–555. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH Value | LogD—Prodrug | LogD—Lazabemide |
|---|---|---|
| 6.4 | –4.58 ± 0.002 | –1.18 ± 0.032 |
| 7.4 | 0.199 ± 0.002 | –0.68 ± 0.019 |
| 7.8 | 0.319 ± 0.003 | –0.63 ± 0.009 |
| Medium | Amount Dissolved (μg/mL) | Amount Measured (μg/mL) |
|---|---|---|
| Water | 6000 | 6277 ± 747 |
| pH 7.4 | 6000 | 6142 ± 730 |
| Concentration of the Test Drug | |||
|---|---|---|---|
| 1 µM | 10 µM | 100 µM | |
| l-Dopa | 102 ± 4.19 | 105 ± 5.24 | 79.3 ± 13.0 |
| l-Dopa–lazabemide prodrug | 112 ± 12.0 | 52.1 ± 14.0 | 34.9 ± 7.94 |
| Permeability (cm/s) Expressed as LogPe | ||||||
|---|---|---|---|---|---|---|
| pH 3.7 | pH 4.7 | pH 5.7 | pH 6.4 | pH 7.4 | pH 7.8 | |
| l-Dopa | −7.99 ± 0.28 | −8.05 ± 0.19 | −8.22 ± 0.14 | −8.62 ± 0.87 | −8.17 ± 0.56 | −8.56 ± 0.07 |
| Prodrug | −8.02 ± 0.39 | −7.81 ± 0.18 | −7.71 ± 0.14 | −7.71 ± 0.06 | −7.53 ± 0.04 | −7.33 ± 0.30 |
| Lazabemide | −7.37 ± 0.24 | −7.06 ± 0.05 | −6.83 ± 0.18 | −6.45 ± 0.28 | −5.79 ± 0.03 | −5.67 ± 0.06 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoon, M.; Petzer, J.P.; Viljoen, F.; Petzer, A. The Design and Evaluation of an l-Dopa–Lazabemide Prodrug for the Treatment of Parkinson’s Disease. Molecules 2017, 22, 2076. https://doi.org/10.3390/molecules22122076
Hoon M, Petzer JP, Viljoen F, Petzer A. The Design and Evaluation of an l-Dopa–Lazabemide Prodrug for the Treatment of Parkinson’s Disease. Molecules. 2017; 22(12):2076. https://doi.org/10.3390/molecules22122076
Chicago/Turabian StyleHoon, Monique, Jacobus P. Petzer, Francois Viljoen, and Anél Petzer. 2017. "The Design and Evaluation of an l-Dopa–Lazabemide Prodrug for the Treatment of Parkinson’s Disease" Molecules 22, no. 12: 2076. https://doi.org/10.3390/molecules22122076
APA StyleHoon, M., Petzer, J. P., Viljoen, F., & Petzer, A. (2017). The Design and Evaluation of an l-Dopa–Lazabemide Prodrug for the Treatment of Parkinson’s Disease. Molecules, 22(12), 2076. https://doi.org/10.3390/molecules22122076