Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers



Abstract
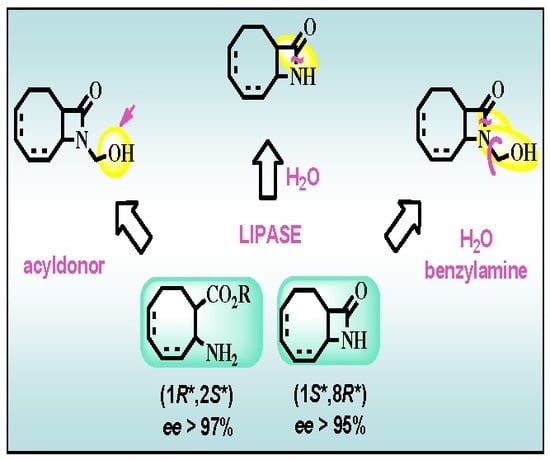
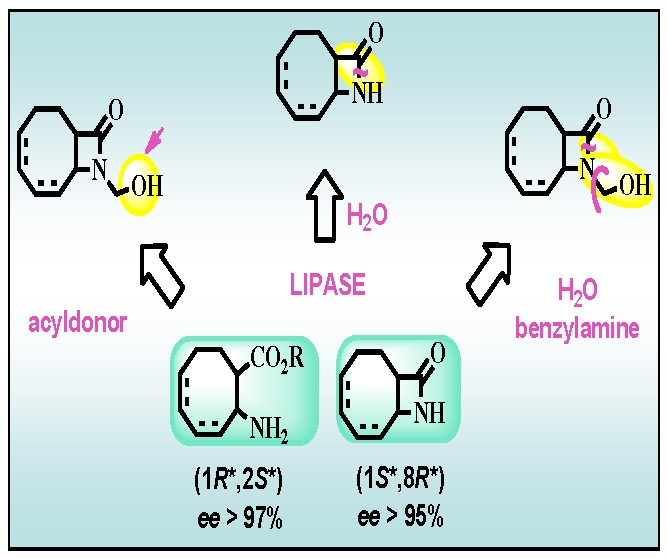
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of β-lactams (±)-3–(±)-6
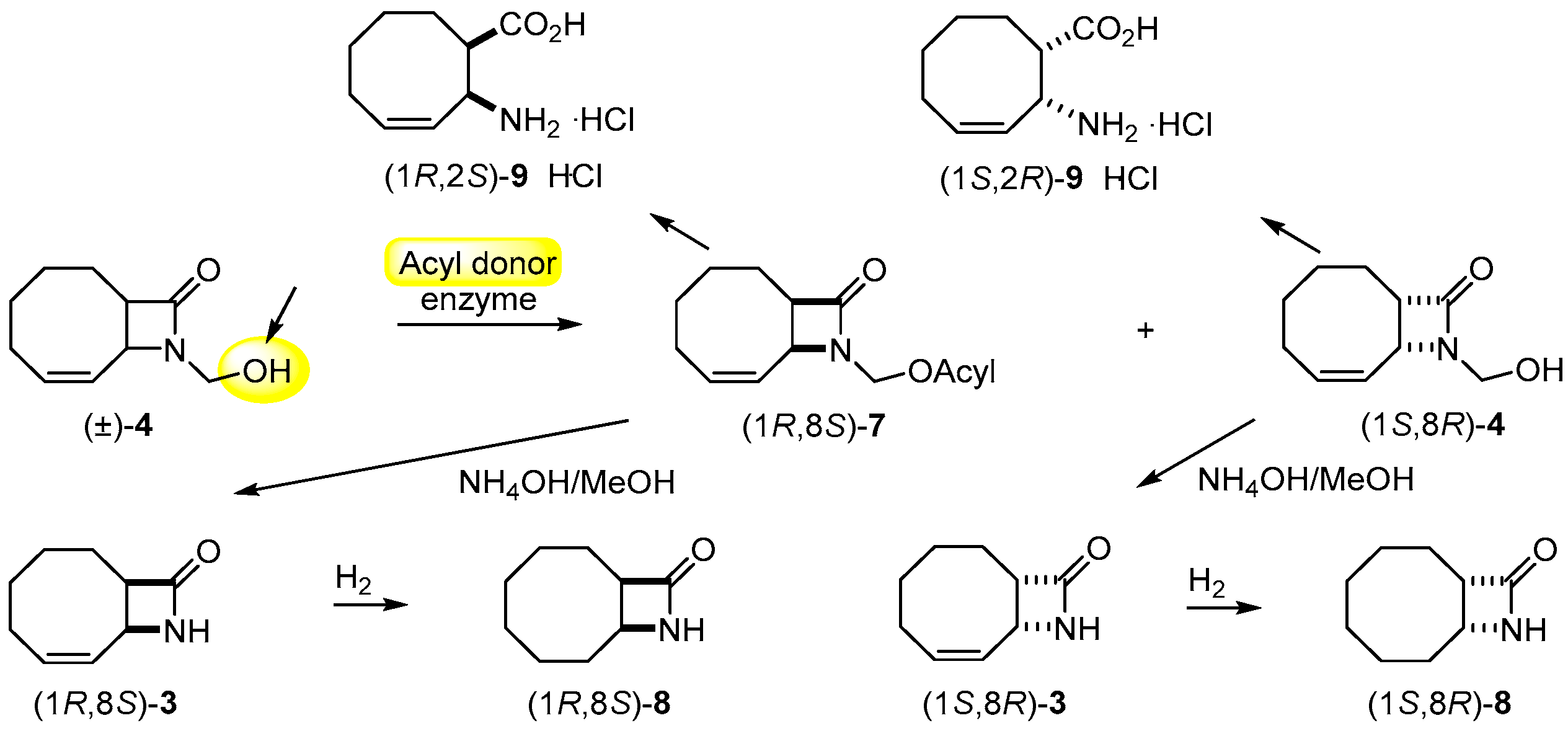
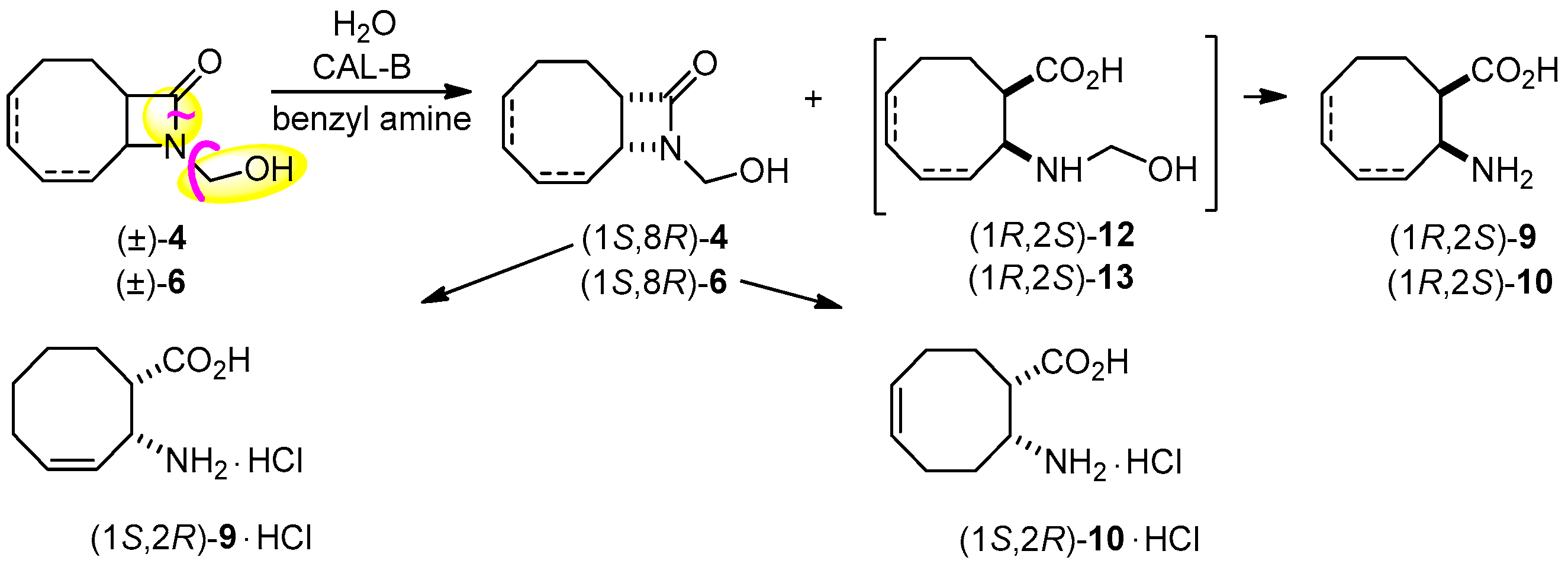
2.2. Lipase-Catalyzed O-acylation of (±)-4
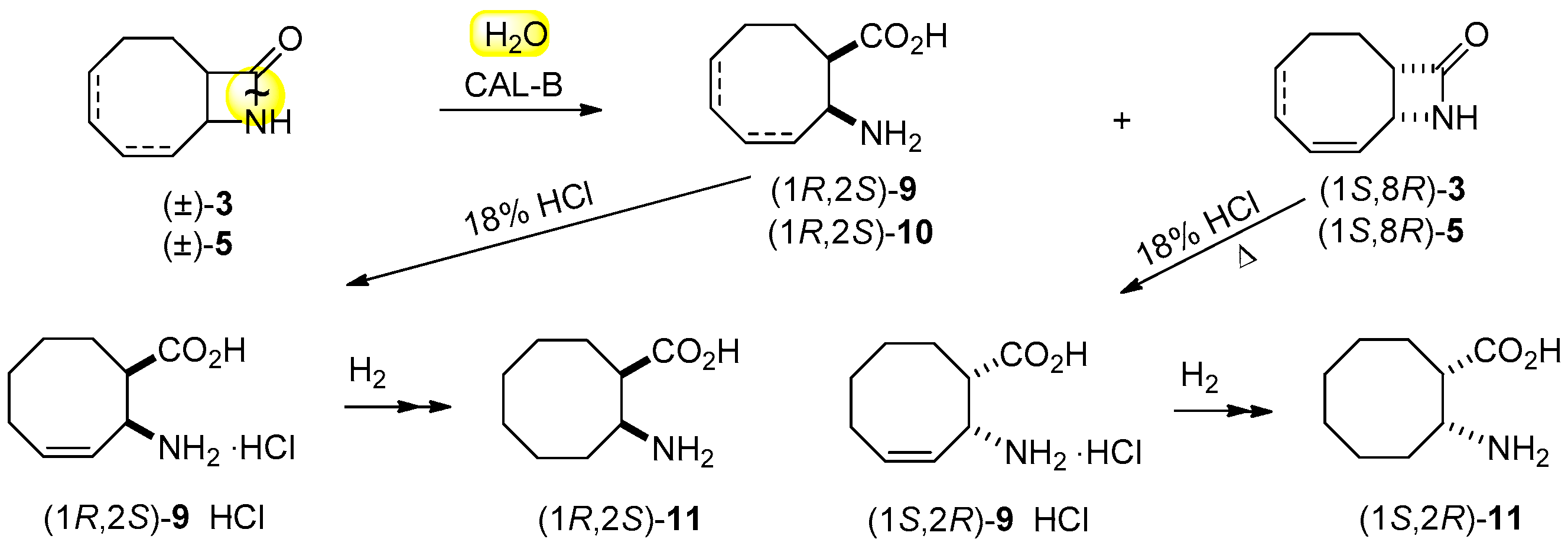
2.3. Lipase-Catalyzed Ring Cleavage of (±)-3–(±)-6
2.4. Further Transformations
2.5. Absolute Configurations
3. Experimental Section
3.1. Materials and Methods
3.2. Typical Small-Scale Enzymatic Experiments
3.3. Synthesis of Racemic 9-Azabicyclo[6.2.0]dec-6-en-10-one [(±)-3]
3.4. Synthesis of Racemic N-Hydroymethyl-9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4]
3.5. Gram-Scale Resolution of N-hydroxymethyl-9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4] through Acylation
3.6. Gram-Scale Resolution of 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-3] through Hydrolysis
3.7. Gram-Scale Resolution of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-6-en-10-one [(±)-4] through Hydrolysis
3.8. Gram-Scale Resolution of N-hydroxymethyl 9-azabicyclo[6.2.0]dec-4-en-10-one [(±)-6] through Hydrolysis
3.9. Synthesis of (1R,8S)-8 and (1S,8R)-8 through (1R,8S)-3 and (1S,8R)-3 from β-lactams (1S,8R)-4 and (1R,8S)-7
3.10. Preparation of (1R,2S)-11 and (1S,2R)-11
3.11. Acidic Hydrolyses to β-amino Acid Hydrochlorides
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fülöp, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, A.; Hahn, M.G.; Dumic, M.; Mittendorf, J. Alicyclic β-amino acids in medicinal chemistry. Amino Acids 2005, 29, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, J.; Kunisch, F.; Matzke, M.; Militzer, H.C.; Schmidt, A.; Schönfeld, W. Novel Antifungal β-Amino Acids: Synthesis and Activity against Candida albicans. Bioorg. Med. Chem. Lett. 2003, 14, 433–436. [Google Scholar] [CrossRef]
- Forró, E.; Árva, J.; Fülöp, F. Preparation of (1R,8S)- and (1S,8R)-9-azabicyclo[6.2.0]dec-4-en-10-one: Potential starting compounds for the synthesis of anatoxin-a. Tetrahedron Asymmetry 2001, 12, 643–649. [Google Scholar] [CrossRef]
- Parsons, P.J.; Camp, N.P.; Edwards, N.; Sumoreeah, L.R. Synthesis of (±)-anatoxin-a and analogues. Tetrahedron 2000, 56, 309–315. [Google Scholar] [CrossRef]
- Wonnacott, S.; Kaiser, S.; Mogg, A.; Soliakov, L.; Jones, I.W. Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. Eur. J. Pharmacol. 2000, 393, 51–58. [Google Scholar] [CrossRef]
- Sharples, C.G.V.; Kaiser, S.; Soliakov, L.; Marks, M.J.; Collins, A.C.; Washburn, M.; Wright, E.; Spencer, J.A.; Gallagher, T.; Whiteaker, P.; et al. UB-165: A Novel Nicotinic Agonist with Subtype Selectivity Implicates the α4β2* Subtype in the Modulation of Dopamine Release from Rat Striatal Synaptosomes. J. Neurosci. 2000, 20, 2783–2791. [Google Scholar] [PubMed]
- Rodriguez, V.; Moura, S.; Pinto, E.; Pereira, C.M.P.; Braga, R.C. Toxicological and chemical aspects of anatoxin-a and its analogs. Quim. Nova 2006, 29, 1465–1471. [Google Scholar]
- Mansell, H.L. Synthetic approaches to anatoxin-a. Tetrahedron 1996, 52, 6025–6061. [Google Scholar] [CrossRef]
- Masato, K.; Shingo, H.; Yasumasa, H.; Tetsuhiro, N. Formal amide insertion strategy for the synthesis of anatoxin-a using rhodium catalysis. Tetrahedron 2016, 72, 1495–1499. [Google Scholar]
- Marc, M.; Outurguin, F.; Renard, P.Y.; Créminon, C.; Franck, X. Synthesis of a (+)-anatoxin-a analogue for monoclonal antibodies production. Tetrahedron Lett. 2009, 50, 4554–4557. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-Amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Mándity, I.M.; Wéber, E.; Martinek, T.A.; Olajos, G.; Tóth, G.K.; Vass, E.; Fülöp, F. Design of peptidic foldamer helices: A stereochemical patterning approach. Angew. Chem. Int. Ed. 2009, 48, 2171–2175. [Google Scholar] [CrossRef] [PubMed]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Miklós, F.; Forró, E. Diexo-3-aminonorbornane-2-carboxylic acid as highly applicable chiral source for the enantioselective synthesis of heterocycles. Synlett 2008, 1687–1689. [Google Scholar] [CrossRef]
- Kazi, B.; Kiss, L.; Forró, E.; Fülöp, F. Synthesis of orthogonally protected azepane β-amino ester enantiomers. Tetrahedron Lett. 2010, 51, 82–85. [Google Scholar] [CrossRef]
- Kanizsai, I.; Gyónfalvi, S.; Szakonyi, Z.; Sillanpää, R.; Fülöp, F. Synthesis of bi- and tricyclic beta-lactam libraries in aqueous medium. Green Chem. 2007, 9, 357–360. [Google Scholar] [CrossRef]
- Liljeblad, A.; Kanerva, L.T. Biocatalysis as a profound tool in the preparation of highly enantiopure beta-amino acids. Tetrahedron 2006, 62, 5831–5854. [Google Scholar] [CrossRef]
- Busto, E.; Gotor-Fernande, V.; Gotor, V. Hydrolases in the stereoselective synthesis of N-heterocyclic amines and amino acid derivatives. Chem. Rev. 2011, 111, 3998–4035. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, M.; Tabassum, R.; Ahmad, M.M.; Hassan, N.A.; Oku, H.; Rivera, G. Enantioselective synthesis of β-amino acids: A Review. Med. Chem. 2015, 5, 295–309. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Direct and indirect enzymatic methods for the preparation of enantiopure cyclic β-amino acids and derivatives from β-lactams. Mini Rev. Org. Chem. 2004, 1, 93–102. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Recent lipase-catalyzed hydrolytic approaches to pharmacologically important β-and γ-amino acids. Curr. Med. Chem. 2012, 19, 6178–6187. [Google Scholar] [PubMed]
- Forró, E.; Fülöp, F. Cispentacin, enzymatic highlights of its 25-years history. Mini Rev. Org. Chem. 2016, 14, 219–226. [Google Scholar] [CrossRef]
- Gyarmati, Z.C.; Liljeblad, A.; Rintola, M.; Bernáth, G.; Kanerva, L.T. Lipase-catalyzed kinetic resolution of 7-, 8- and 12-membered alicyclic β-amino esters and N-hydroxymethyl-β-lactam enantiomers. Tetrahedron Asymmetry 2003, 14, 3805–3814. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. Lipase-catalyzed enantioselective ring opening of unactivated alicyclic-fused β-lactams in an organic solvent. Org. Lett. 2003, 5, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Fülöp, F. Advanced procedure for the enzymatic ring opening of unsaturated alicyclic β-lactams. Tetrahedron Asymmetry 2004, 15, 2875–2880. [Google Scholar] [CrossRef]
- Forró, E.; Fülöp, F. An efficient enzymatic synthesis of benzocispentacin and its new six- and seven-membered homologues. Chem. Eur. J. 2006, 12, 2587–2592. [Google Scholar] [CrossRef] [PubMed]
- Forró, E.; Galla, Z.; Fülöp, F. The N-hydroxymethyl group as a traceless activating group for the CAL-B-catalysed ring cleavage of β-lactams: A type of two-Step cascade reaction. Eur. J. Org. Chem. 2016, 2647–2652. [Google Scholar] [CrossRef]
- Nativ, E.; Rona, P. 2-Azabicyclo [3.2.0] heptane-3-one. Isr. J. Chem. 1972, 10, 55–58. [Google Scholar] [CrossRef]
- Singh, R.; Cooper, R.D.G. Synthesis and biological evaluation of 6-azabicyclo [3.2.0] hept-2-ene derivatives as potential anti-bacterial agents and β-lactamase inhibitors. Tetrahedron 1994, 50, 12049–12064. [Google Scholar] [CrossRef]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a quick double-derivatization technique. J. Chromatogr. A. 2009, 1216, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors in mg quantities. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme (30 mg mL−1) | Acyl Donor (Equiv) | Solvent | Temp. (°C) | R. Time (Min) | Conv. (%) | ees b (%) | eep b (%) | E |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PSIM | VB (2) | iPr2O | -15 | 120 | 44 | 76 | 96 | 112 |
| 2 | PSIM | VB (2) | iPr2O | 2-3 | 60 | 43 | 70 | 94 | 67 |
| 3 | PSIM | VB (2) | iPr2O | 30 | 10 | 46 | 77 | 90 | 44 |
| 4 | PSIM | VB (10) | iPr2O | 30 | 10 | 51 | 87 | 84 | 32 |
| 5 | PSIM | VB (10) + Et3N + Na2SO4 | iPr2O | 30 | 10 25 | 45 50 | 77 91 | 96 92 | 114 76 |
| 6 | PSIM | 2,2,2-Trifluoroethyl-butyrate(10) | iPr2O | 30 | 20 | 49 | 83 | 86 | 34 |
| 7 | PSIM | VA (10) | iPr2O | 30 | 10 | 50 | 81 | 82 | 25 |
| 8 | PSIM | EtOAc(10) | iPr2O | 30 | 240 | 16 | 12 | 62 | 5 |
| 9 | PSIM | Ac2O (10) | iPr2O | 30 | 10 | 52 | 87 | 80 | 25 |
| 10 | AK c | VB (10) | iPr2O | 30 | 20 | 49 | 74 | 78 | 17 |
| 11 | AY c | VB (10) | iPr2O | 30 | 240 | 25 | 14 | 42 | 28 |
| 12 | CAL-A c | VB (10) | iPr2O | 30 | 10 | 29 | 12 | 29 | 2 |
| 14 | CAL-B | VB (10) | iPr2O | 30 | 50 | 67 | 2 | 1 | 1 |
| 14 | PPL | VB (10) | iPr2O | 30 | 120 | 20 | 23 | 91 | 26 |
| 15 | PSIM | VB (10) | tBuOMe | 30 | 10 | 51 | 87 | 85 | 34 |
| 16 | PSIM | VB (10) | Toluene | 30 | 5 | 49 | 89 | 93 | 82 |
| 17 | PSIM | VB (10) | Acetone | 30 | 60 | 49 | 86 | 89 | 47 |
| Entry | Substrate | R. Time (h) | Conv. c (%) | ees d (%) | eep e (%) |
|---|---|---|---|---|---|
| 1 | (±)-3 | 5 | 43 | 75 | >99 |
| 2 | (±)-5 | 5 | 46 | 84 | >99 |
| 3 | (±)-4 | 3 | 49 | 96 | >99 |
| 4 | (±)-6 | 3 | 50 | >99 | 98 |
| Product | Unreacted substrate | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Reaction Partner | R. Time (Min) | Conv. a (%) | Yield (%) | Isomer | eeP (%) | Yield (%) | Isomer | eeS (%) | (EtOH) | ||
| (±)-3 b | H2O | 330 | 50 | 48 | (1R,2S)-9 | 99 c | −17 d | 49 | (1S,8R)-3 | 99 e | −140.6 f |
| (±)-4 g | VB | 10 | 51 | 46 | (1R,8S)-7 | 94 e | +39.2 h | 44 | (1S,8R)-4 | 96 e | −142.4 i |
| (±)-4 j | H2O | 180 | 49 | 47 | (1R,2S)-9 | >99 c | −17.1 d | 48 | (1S,8R)-4 | 98 e | −140.4 f |
| (±)-6 j | H2O | 180 | 50 | 47 | (1R,2S)-10 | 99 c | +24.9 k | 46 | (1S,8R)-6 | 99 e | −28.7 l |
| Entry | Enantiomers | ee (%) | |
|---|---|---|---|
| 1 | (1R,8S) 3 from (1R,8S)-7 | 95 | +147 (c = 0.5; EtOH) |
| 2 | (1S,8R) 3 from (1S,8R)-4 | 96 | −148.7 (c = 0.4; EtOH) |
| 3 | (1R,8S) 8 from (1R,8S)-3 | 98 | +17.7 (c = 0.5; CHCl3) |
| 4 | (1S,8R) 8 from (1S,8R)-3 | 96 | −17.1 (c = 0.5; CHCl3) |
| 5 | (1S,2R)-9·HCl from (1S,8R) 3 | 99 | +19.6 (c = 0.5; H2O) |
| 6 | (1S,2R)-9·HCl from (1S,8R) 4 | 99 | +19.6 (c = 0.6; H2O) |
| 7 | (1R,2S)-9·HCl from (1R,8S)-7 | 98 | −17.3 (c = 0.35; H2O) |
| 8 | (1S,2R)-10·HCl from (1S,8R)-6 | 97 | −15.0 (c = 0.5; H2O) |
| 9 | (1R,2S)-11 from (1R,2S)-9 | 99 | +19.2 (c = 0.4; H2O) |
| 10 | (1S,2R)-11 from (1S,2R)-9 | 99 | −19 (c = 0.33; H2O) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forró, E.; Kiss, L.; Árva, J.; Fülöp, F. Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers. Molecules 2017, 22, 2211. https://doi.org/10.3390/molecules22122211
Forró E, Kiss L, Árva J, Fülöp F. Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers. Molecules. 2017; 22(12):2211. https://doi.org/10.3390/molecules22122211
Chicago/Turabian StyleForró, Enikő, Loránd Kiss, Judit Árva, and Ferenc Fülöp. 2017. "Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers" Molecules 22, no. 12: 2211. https://doi.org/10.3390/molecules22122211
APA StyleForró, E., Kiss, L., Árva, J., & Fülöp, F. (2017). Efficient Enzymatic Routes for the Synthesis of New Eight-membered Cyclic β-Amino Acid and β-Lactam Enantiomers. Molecules, 22(12), 2211. https://doi.org/10.3390/molecules22122211