
The Therapeutic Potential of Migrastatin-Core Analogs for the Treatment of Metastatic Cancer



Abstract
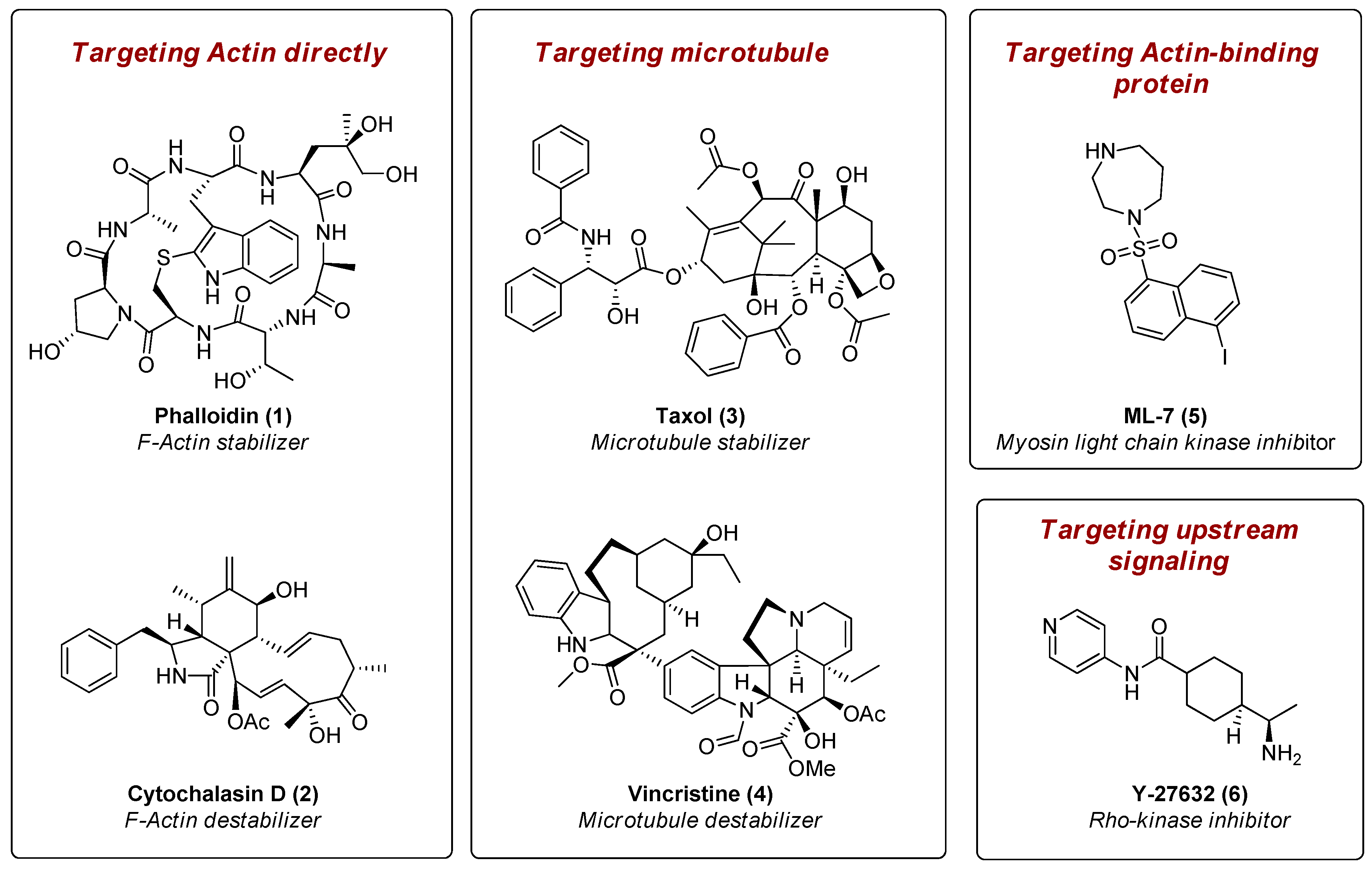
:1. Introduction
2. Results
2.1. Synthesis of Advanced Intermediate 8
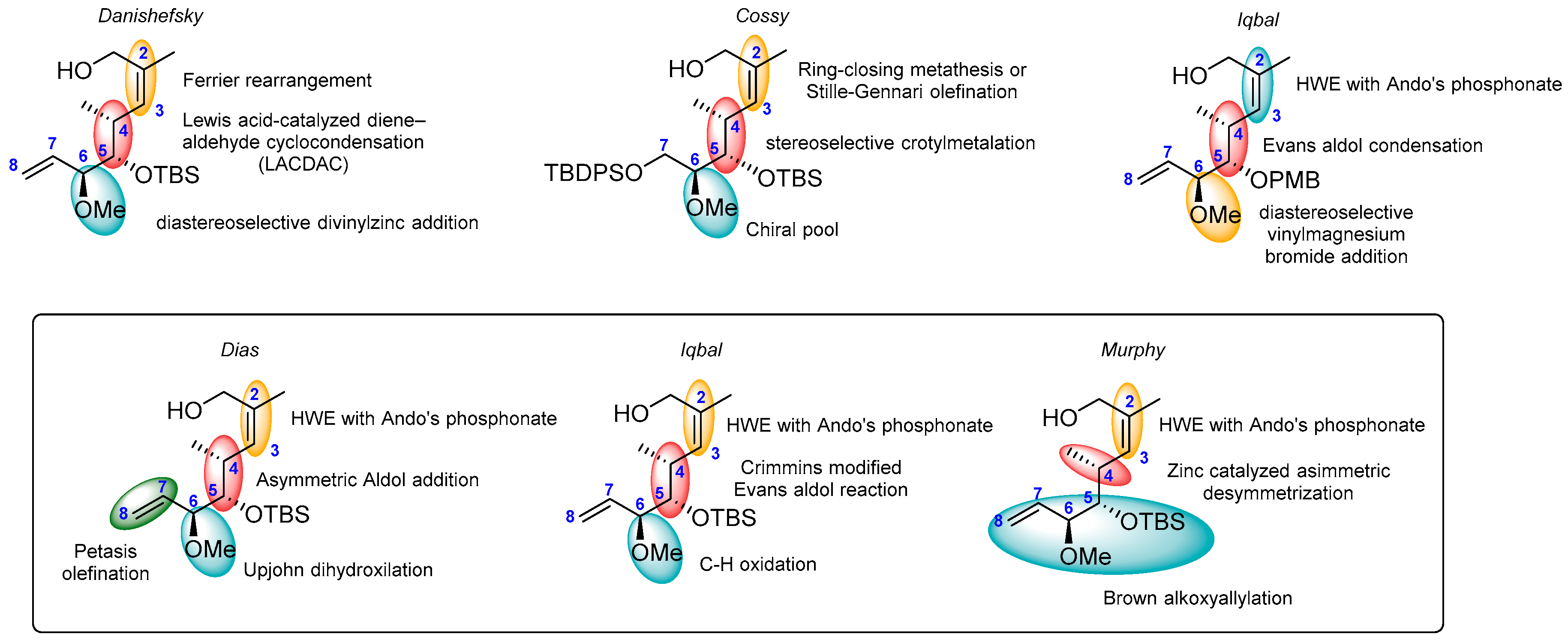
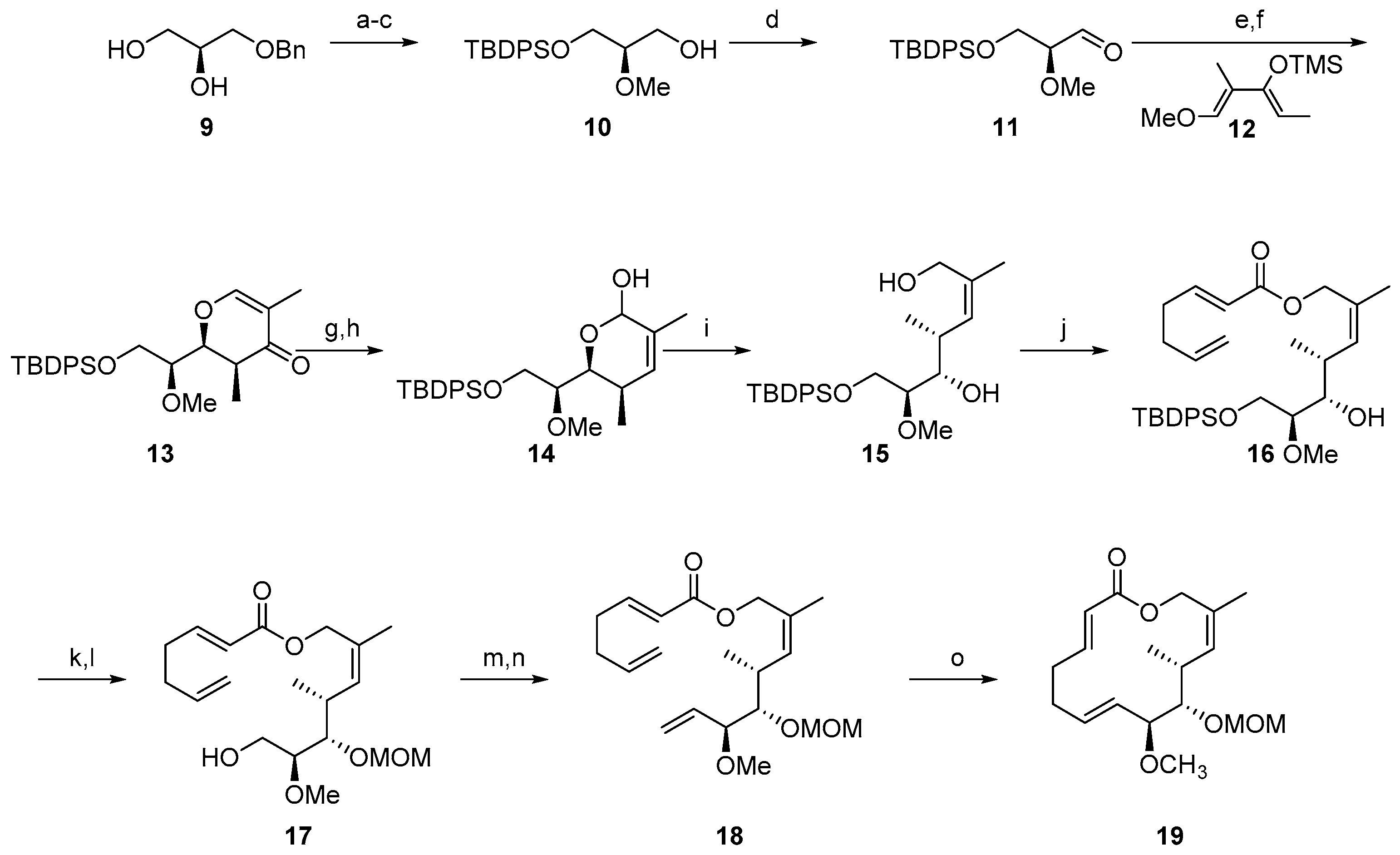
2.1.1. Synthesis of the Protected Migrastatin-Core (Danishefsky et al.) [16]
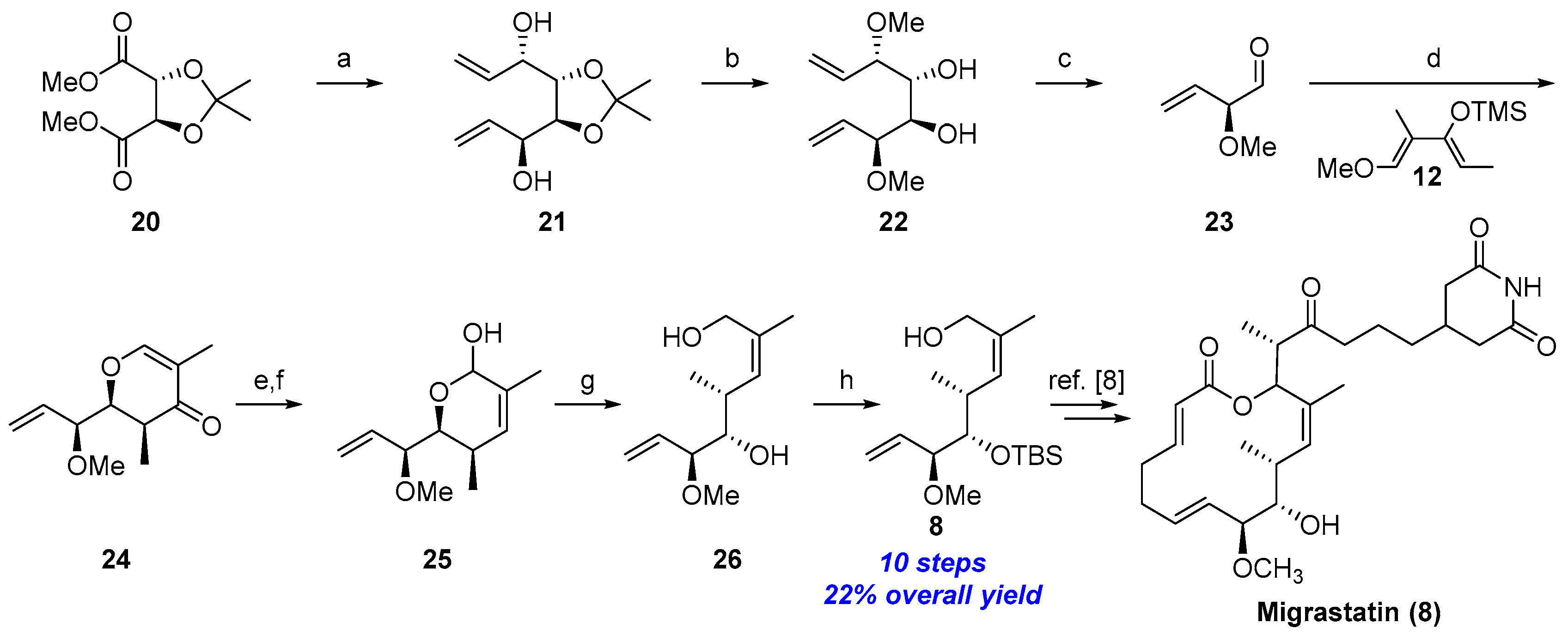
2.1.2. First Total Synthesis of Migrastatin (Danishefsky et al.) [8]
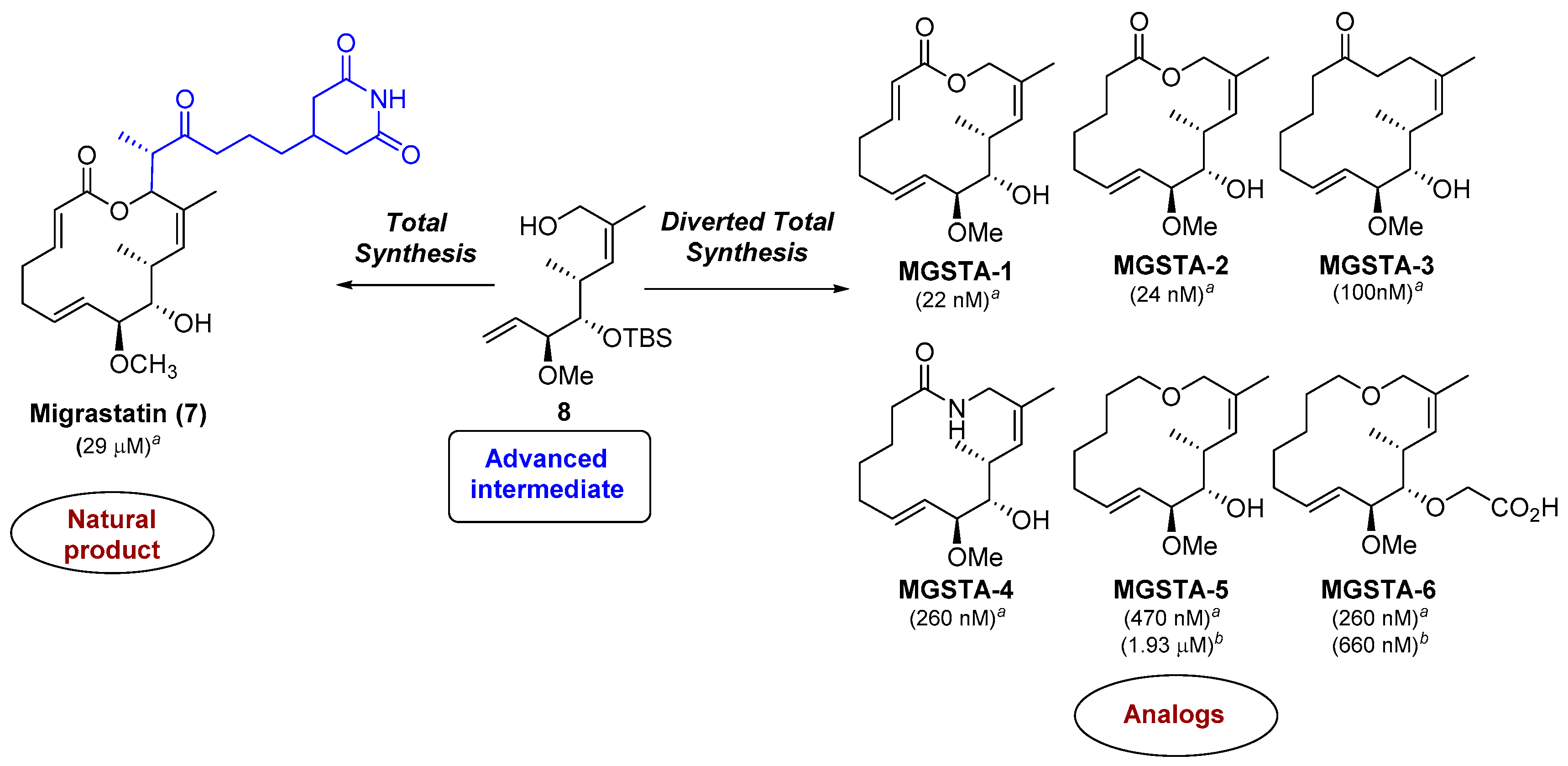
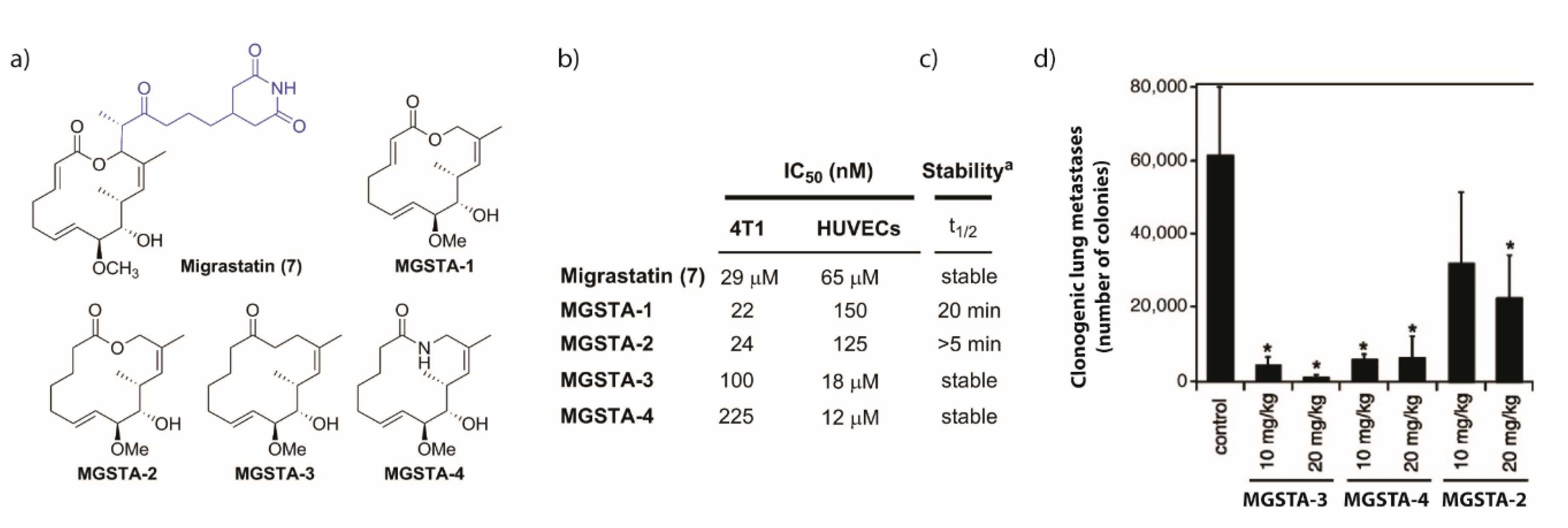
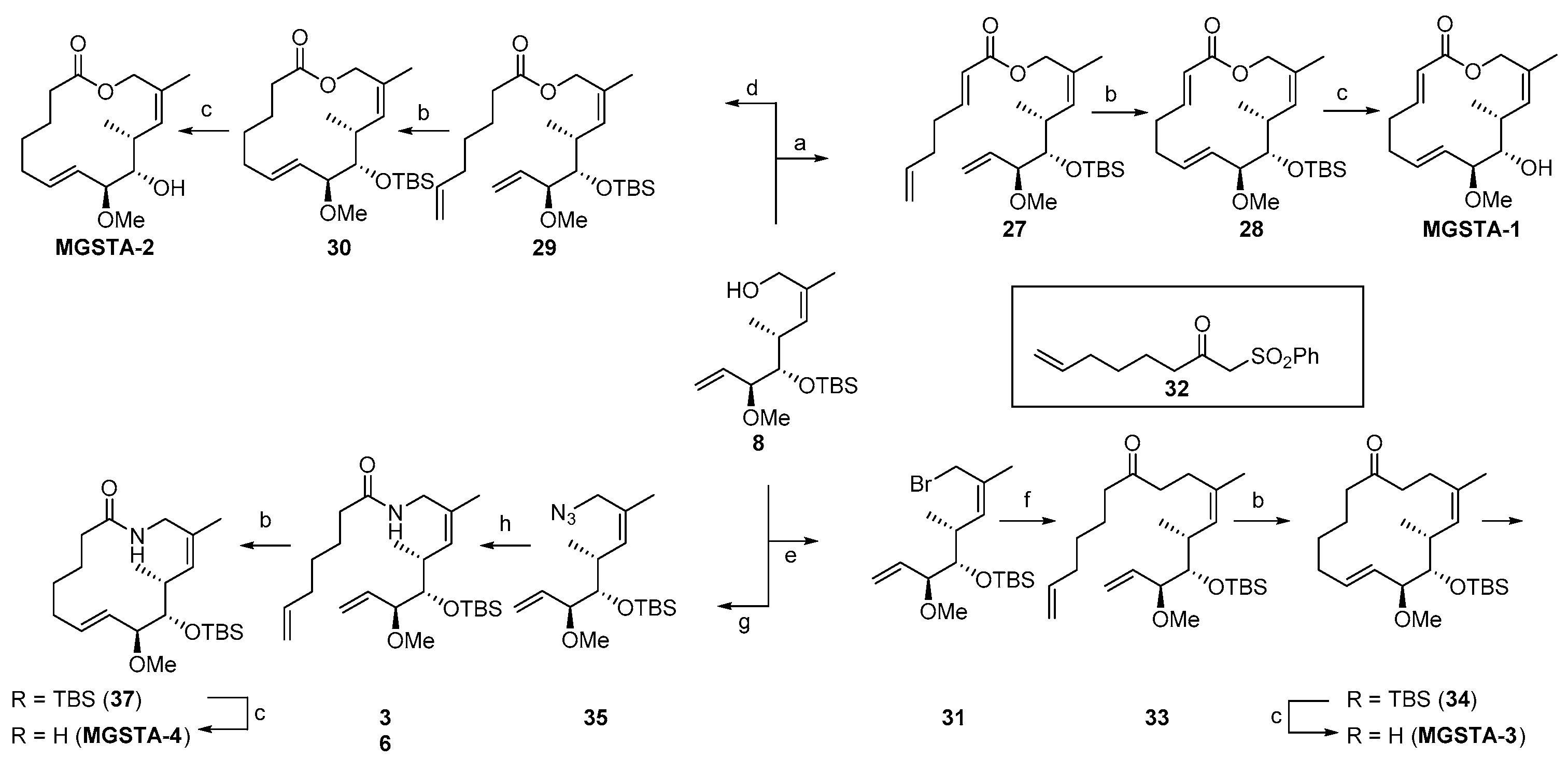
2.1.3. Synthesis of the Migrastatin-Core Library (Danishefsky et al.) [9,29]
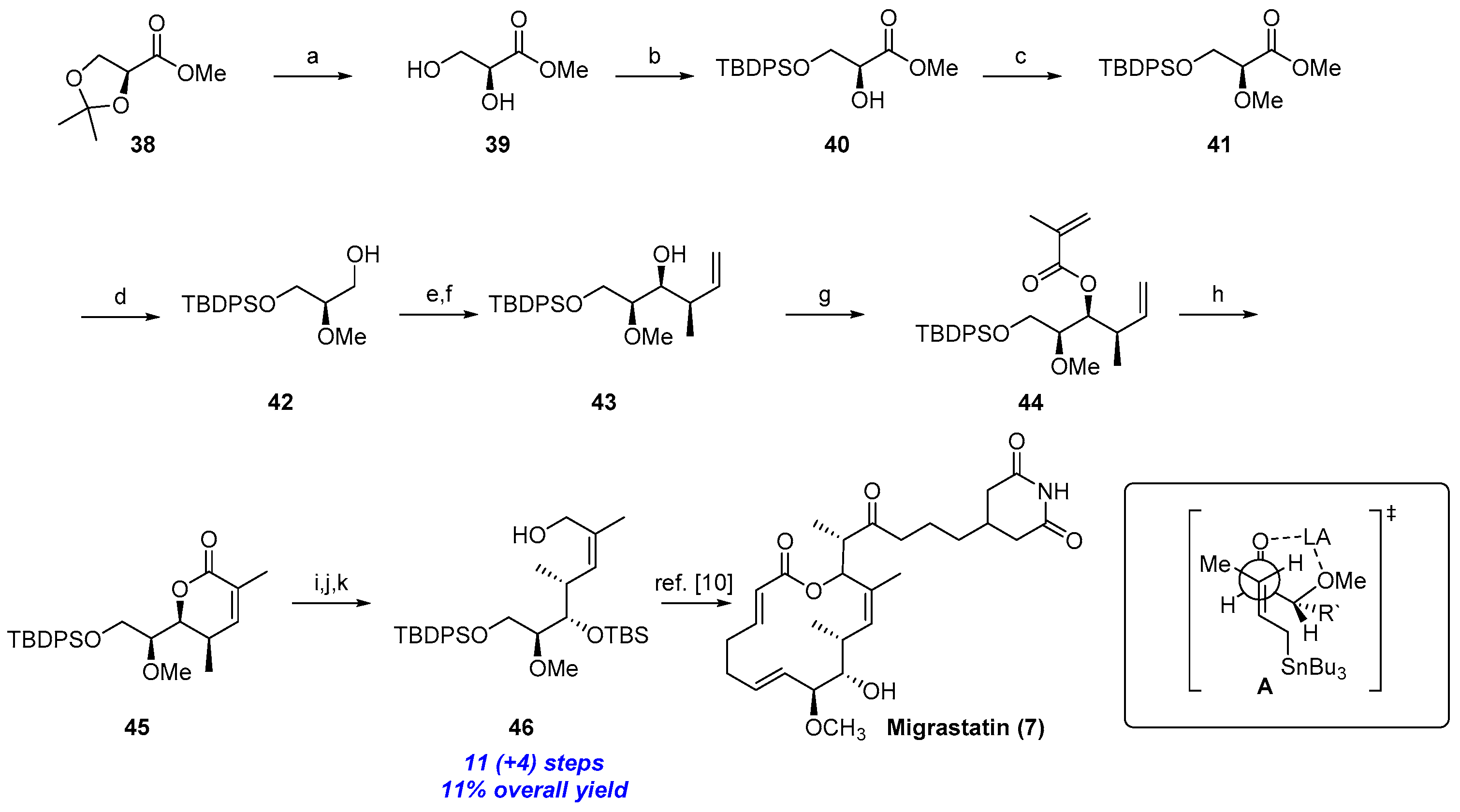
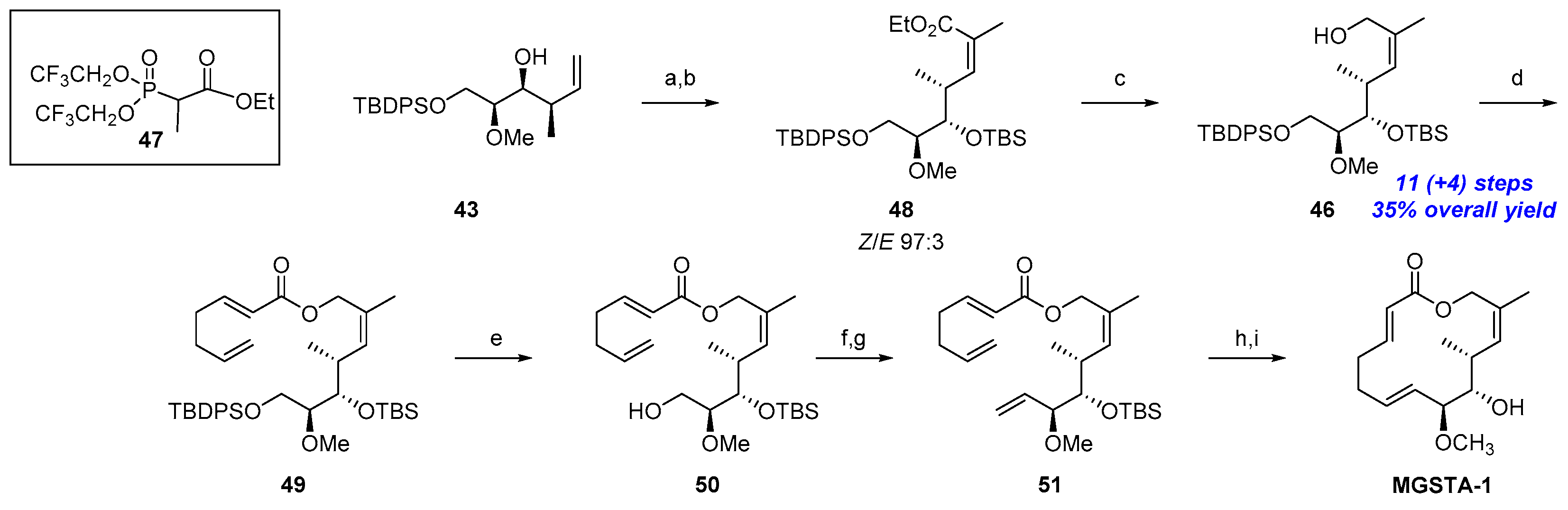
2.1.4. Synthesis by Cossy et al. [10,17]
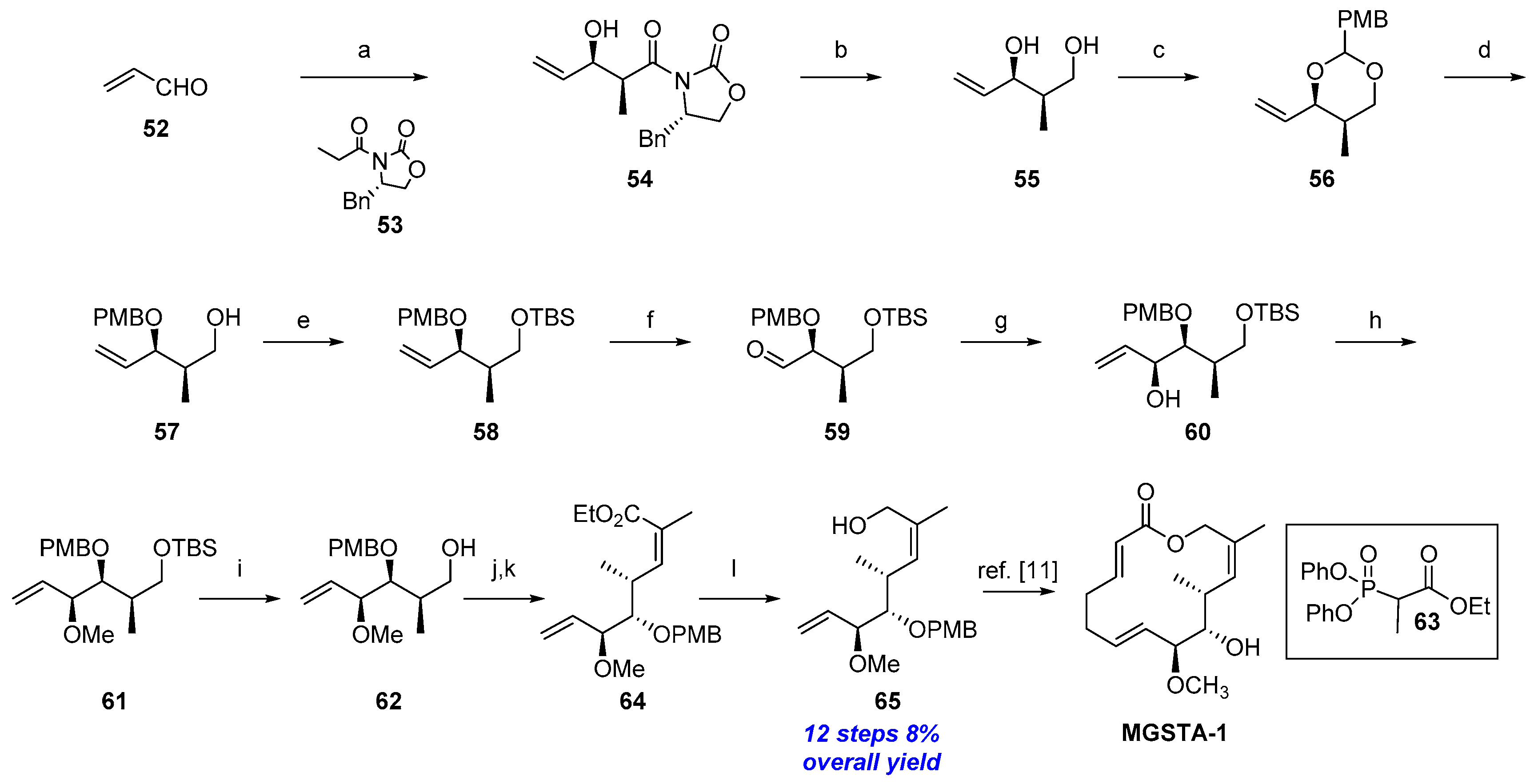
2.1.5. Synthesis by Iqbal et al. [11]
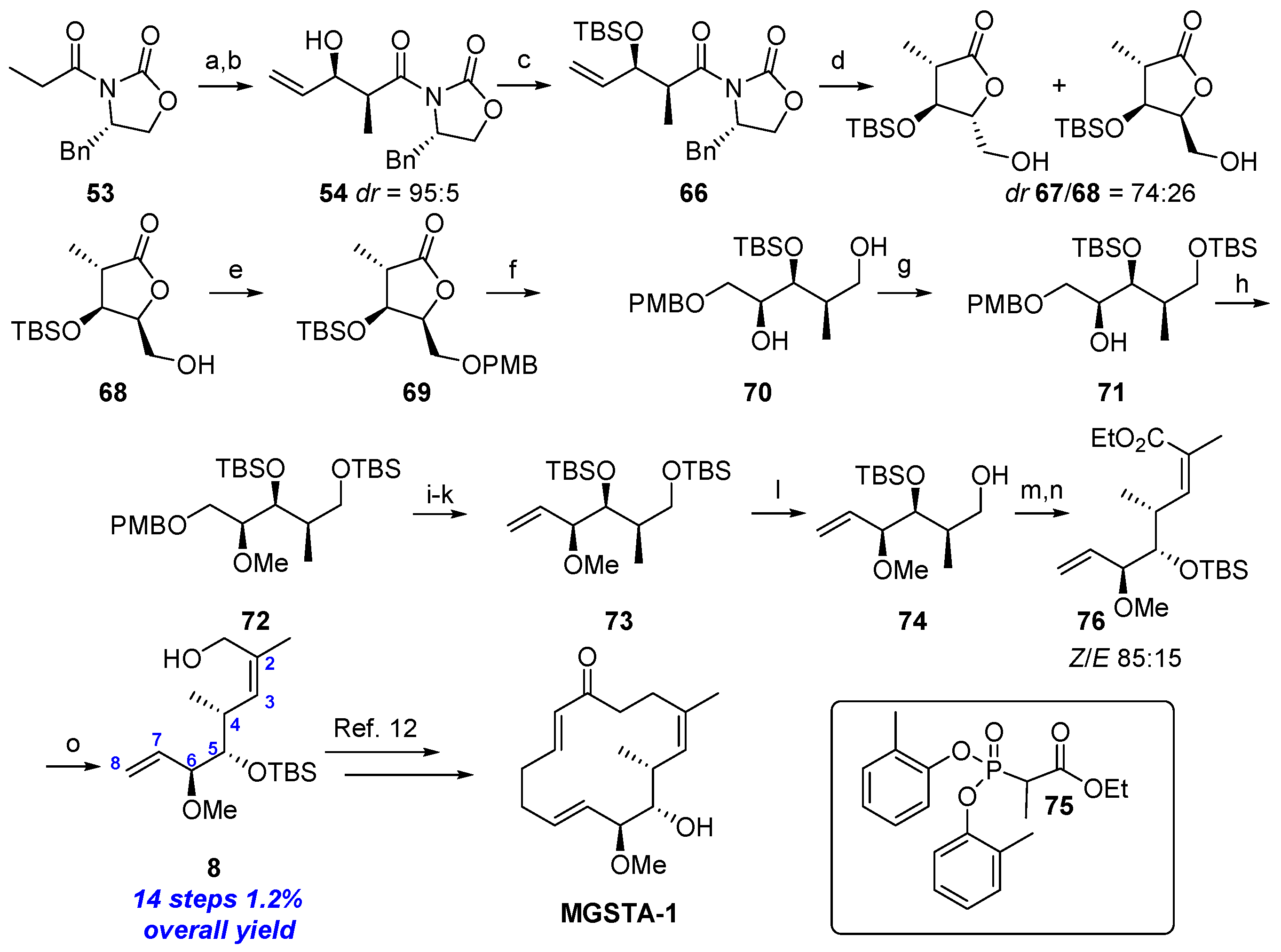
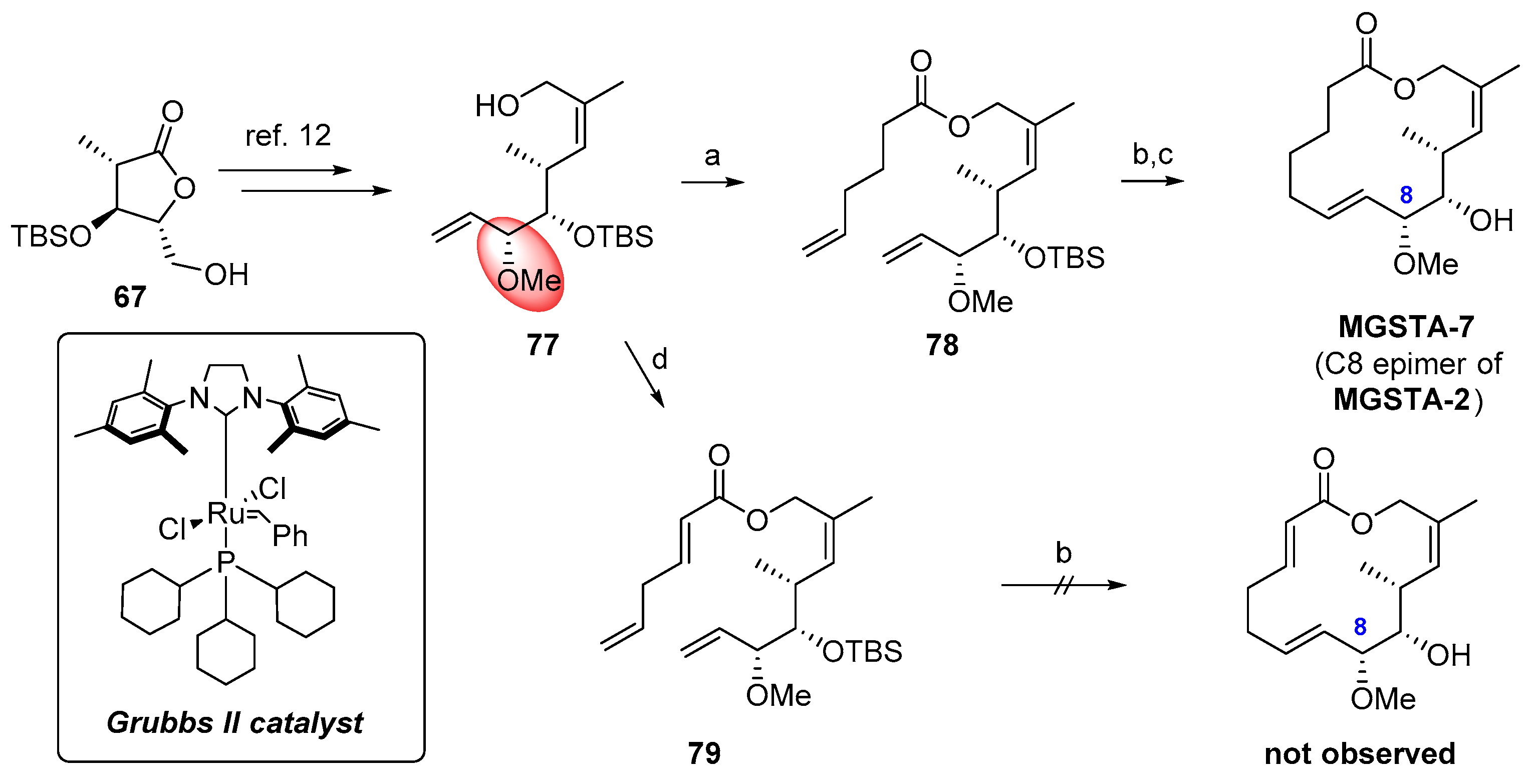
2.1.6. Synthesis by Dias et al. [12]
2.1.7. Synthesis by Iqbal et al. [13]
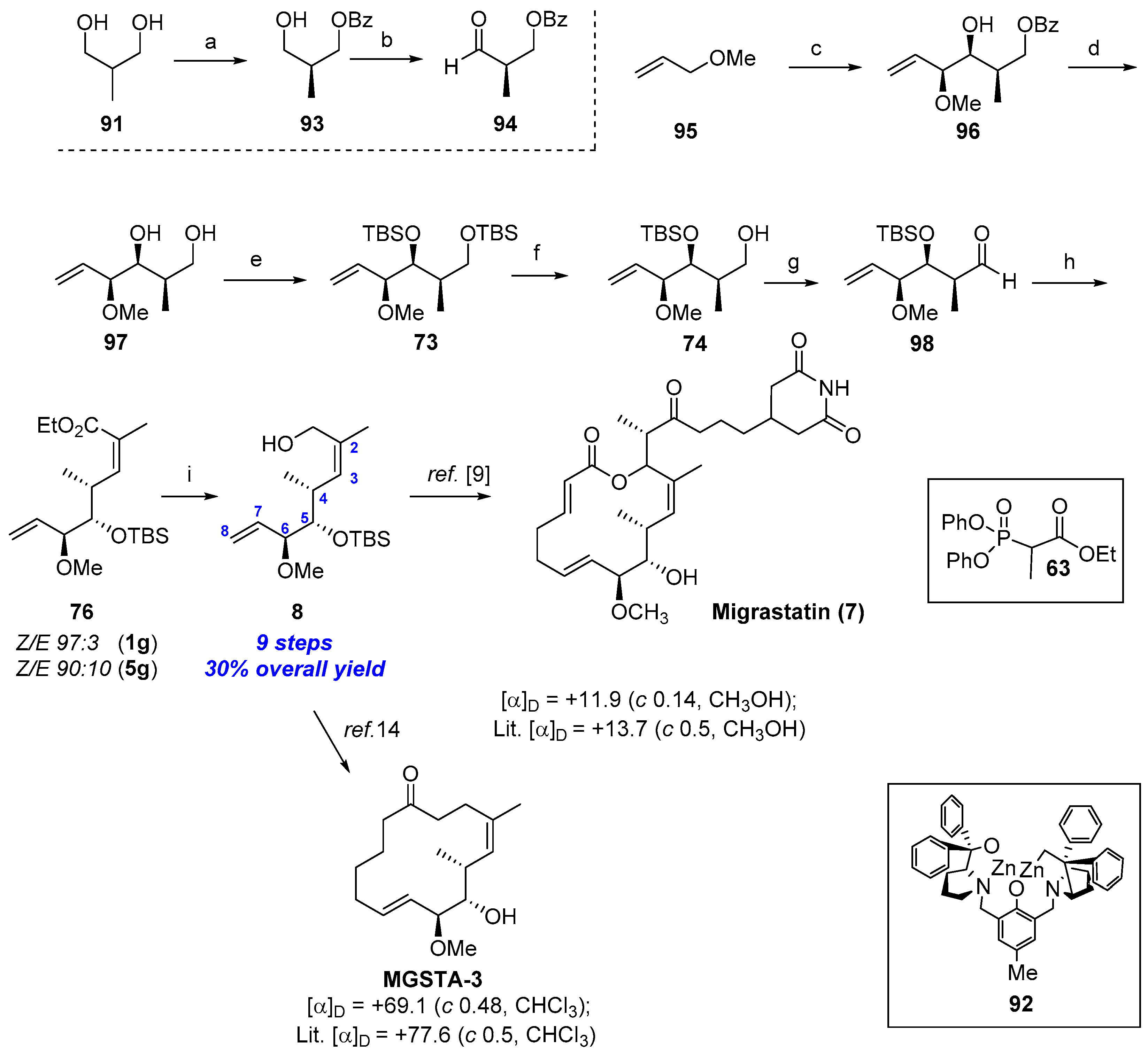
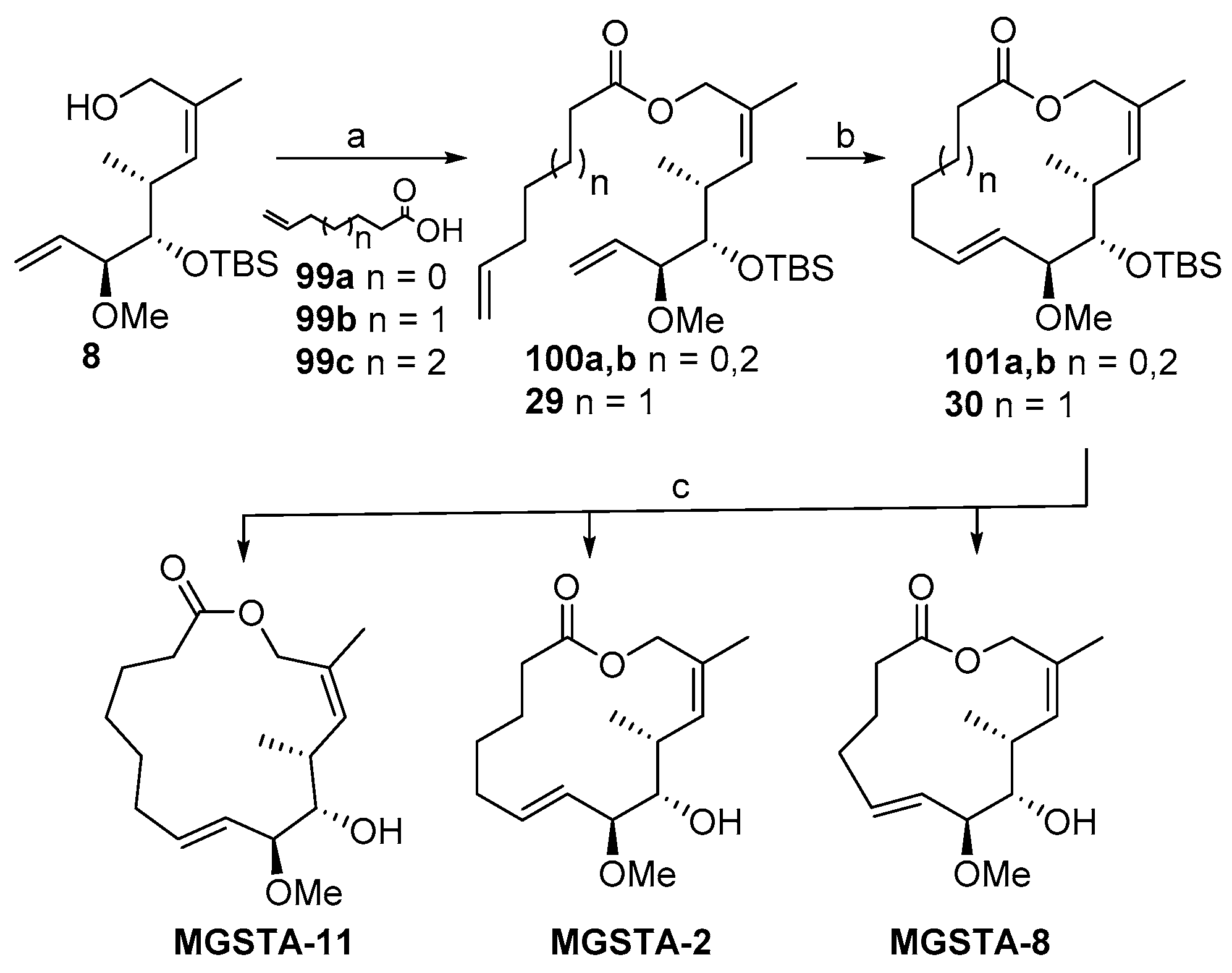
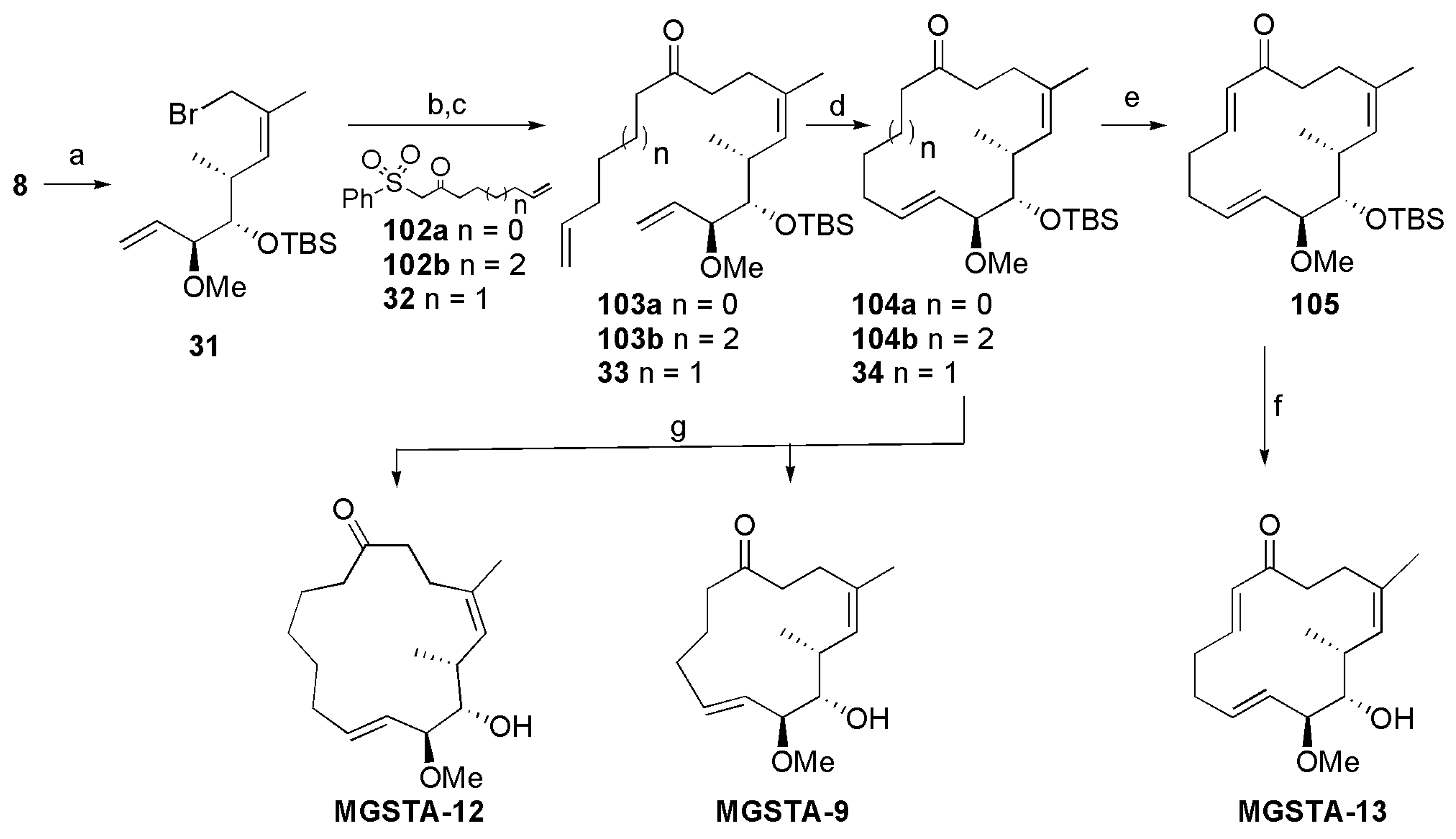
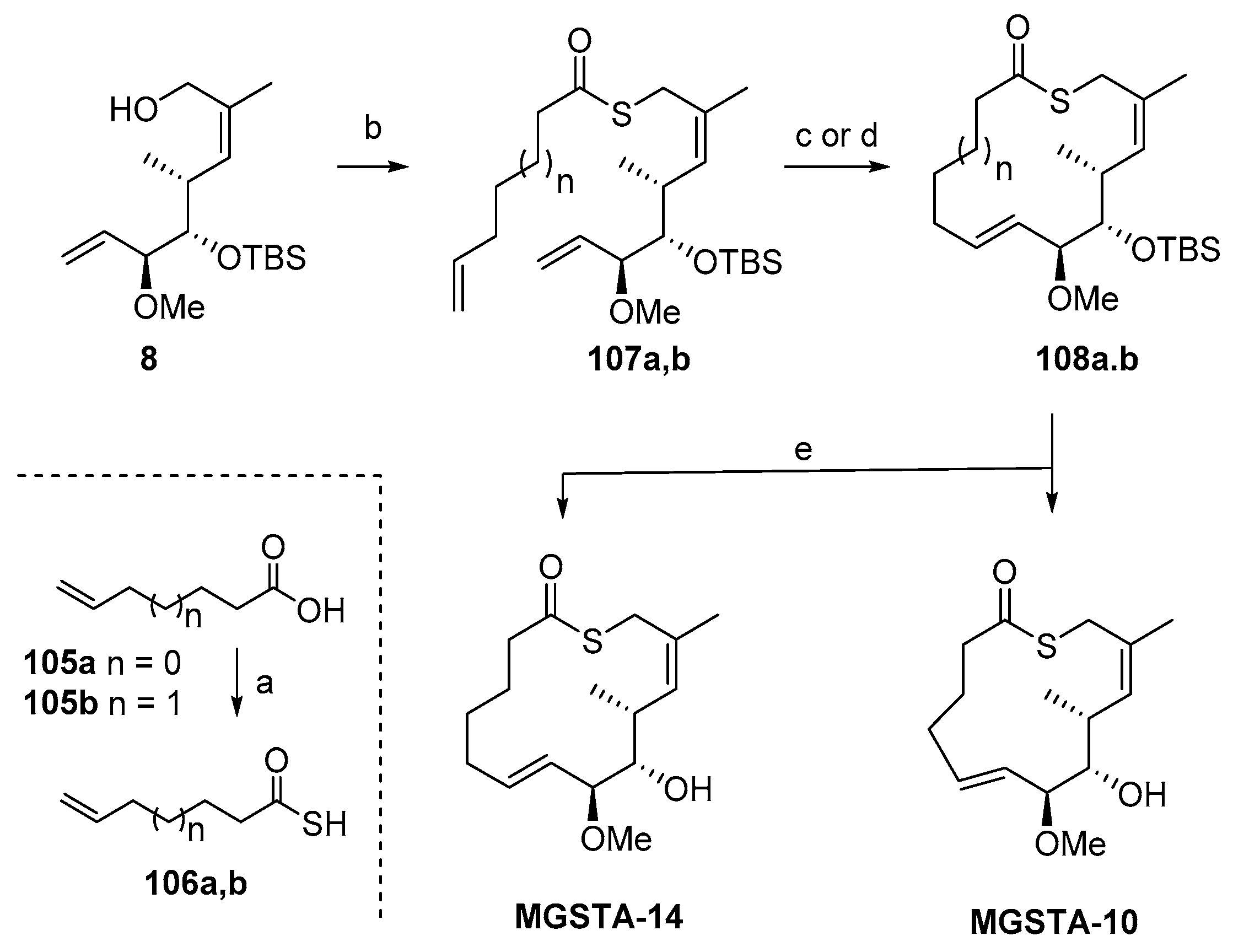
2.1.8. Synthesis by Murphy et al. [14]
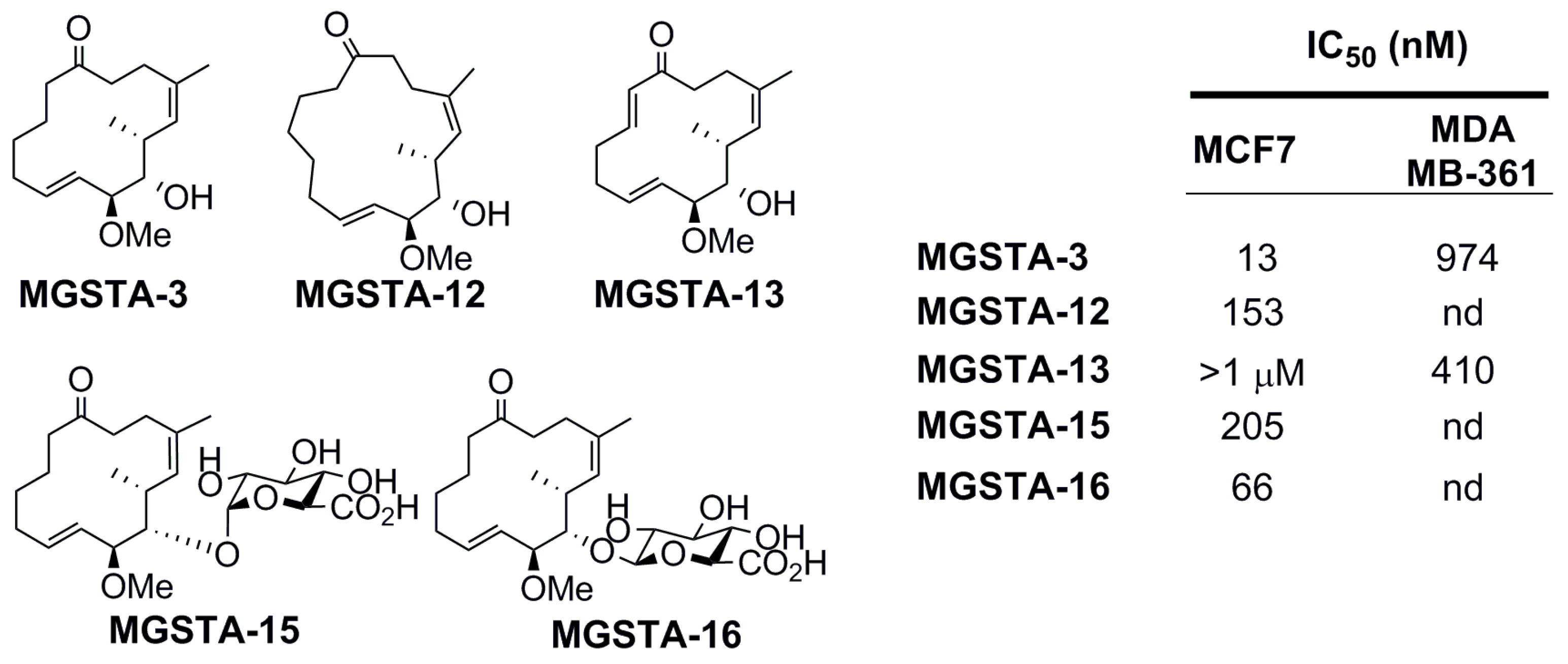
2.2. Biology
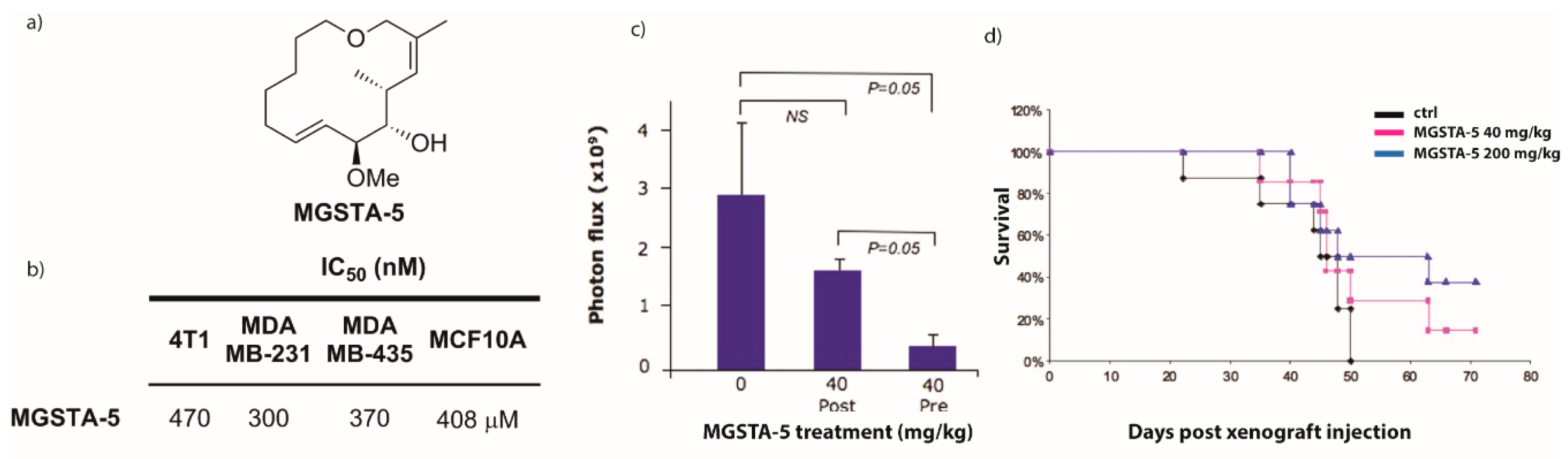
2.2.1. Preliminary Findings [3,6,83]
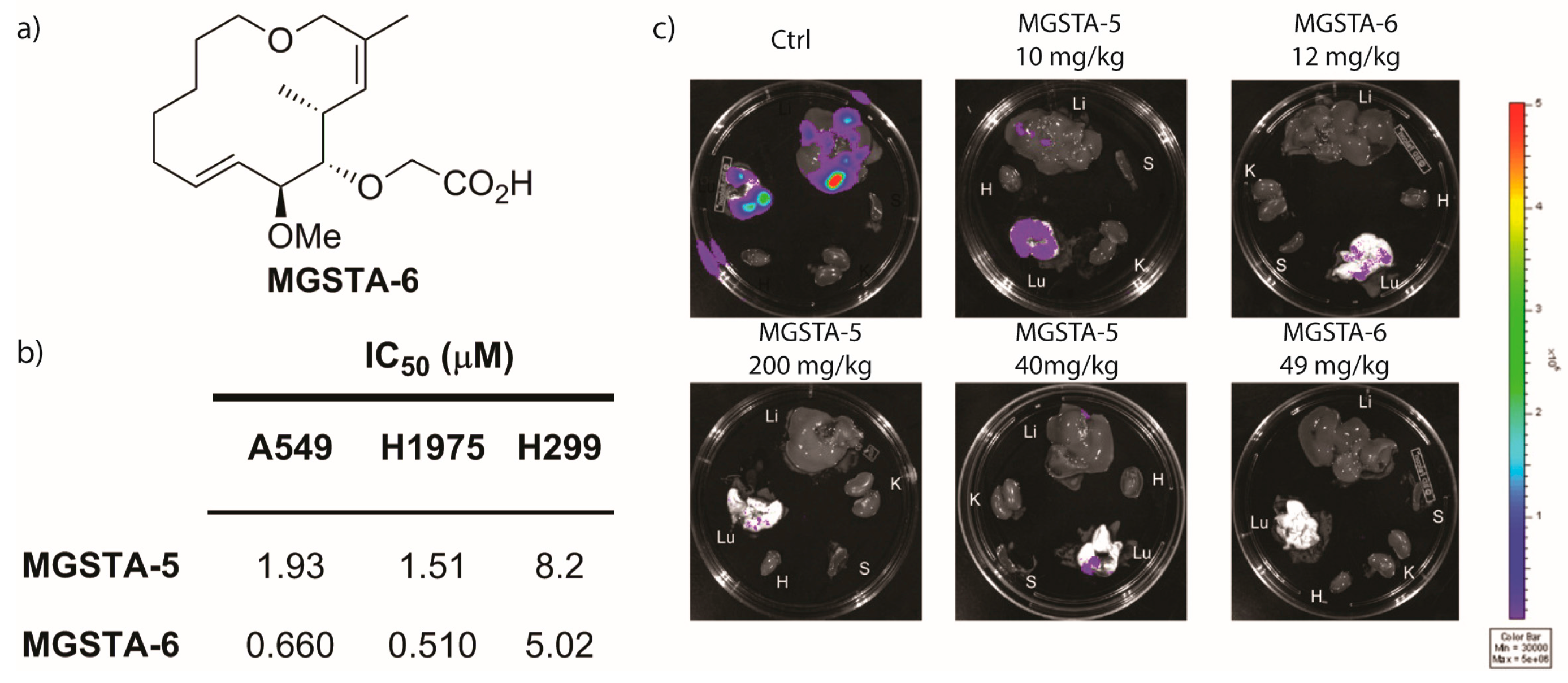
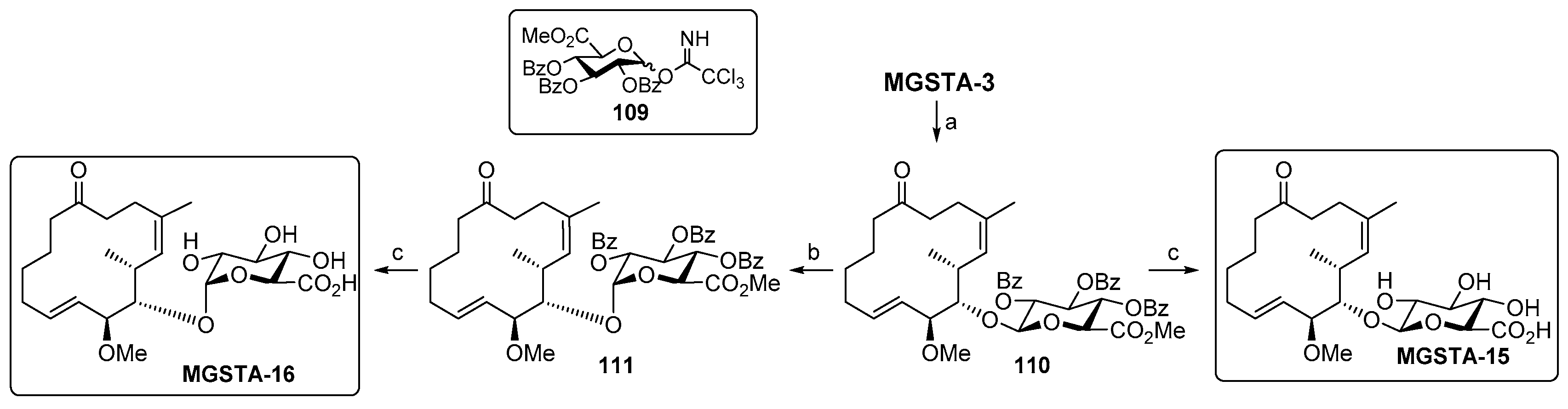
2.2.2. Danishefsky’s Work [9,86,87,88]
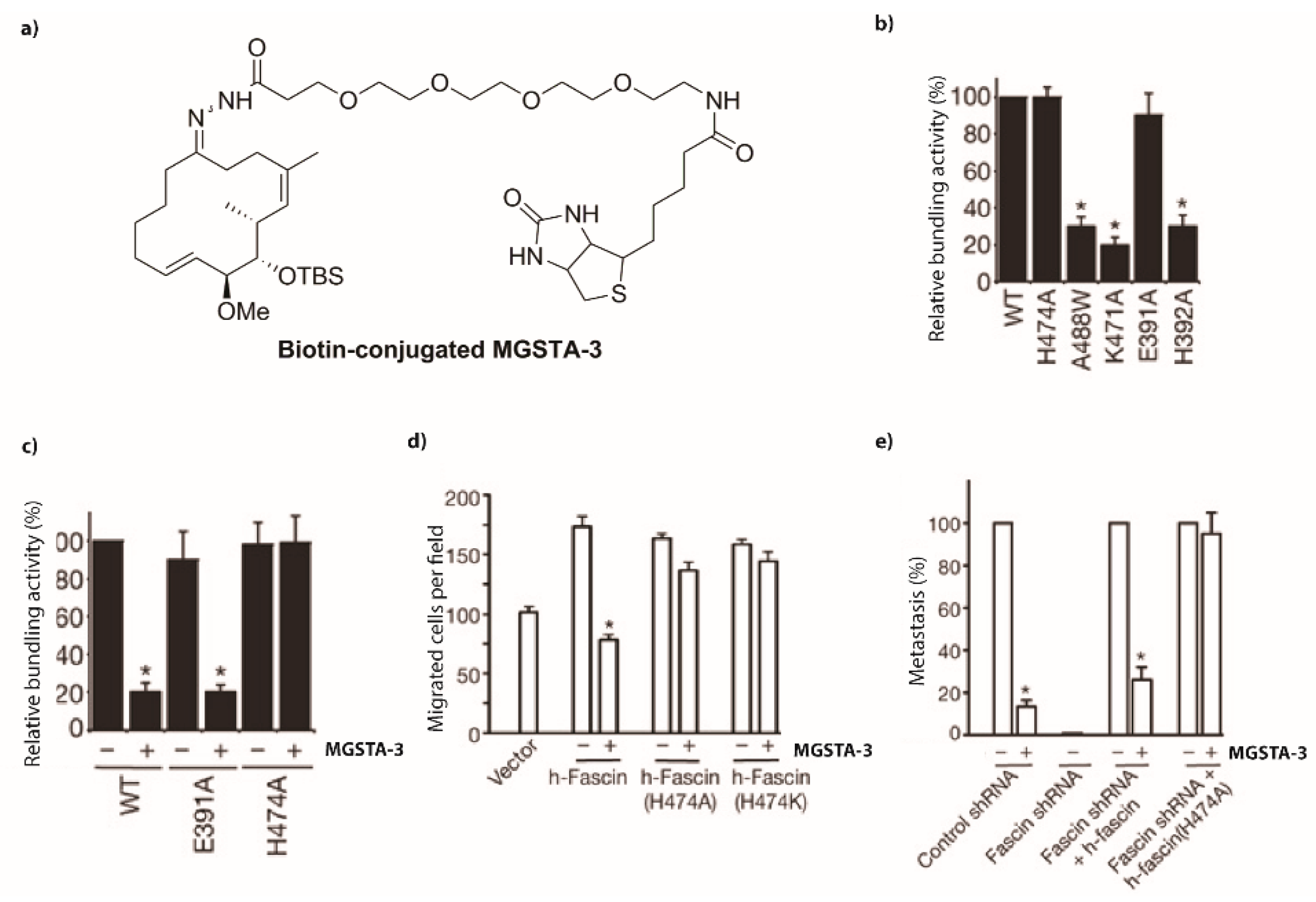
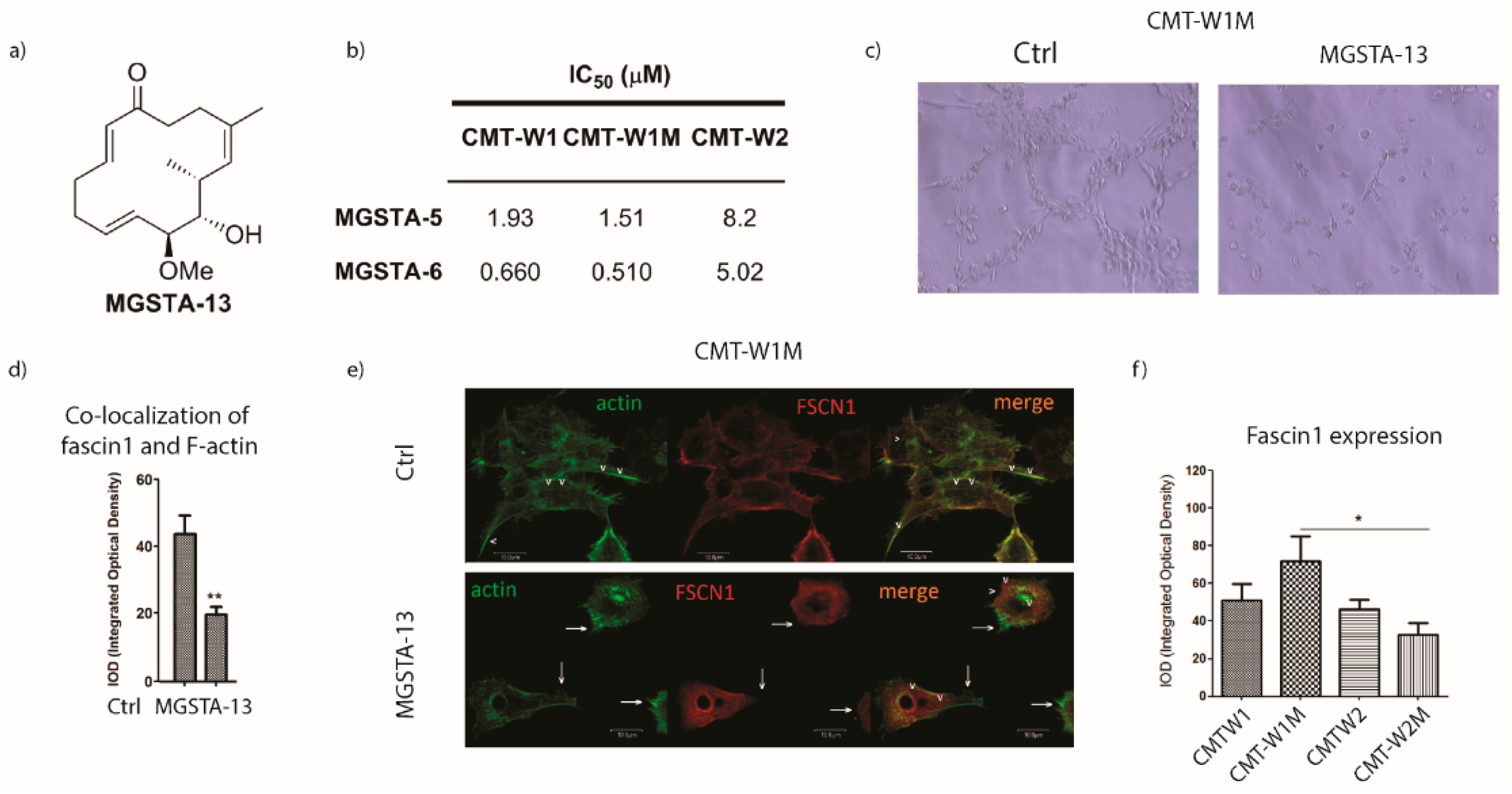
2.2.3. Mechanism of Action [93]
Acknowledgments
Conflicts of Interest
Abbreviations
| 2,6-lutidine | 2,6-dimethylpyridine |
| BAIB | (Diacetoxyiodo) benzene |
| CSA | camphorsulfuric acid |
| DCC | N,N’-dicyclohexylcarbodiimide |
| DIBAL | diisobutylaluminium hydride |
| DIPEA | N,N-Diisopropylethylamine |
| DMP | Dess-Martin periodinane |
| DMAP | 4-dimethylaminopyridine |
| DPPA | diphenylphosphoryl azide |
| DTBMP | 2,6-di-tert-butyl-4-methylpyridine |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Grubbs II | (1,3-Bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(phenylmethylene) (tricyclohexylphosphine) ruthenium |
| (+)Ipc2BOMe | (+)-B-methoxydiisopinocampheylborane |
| MS | molecular sieves |
| MeOTf | methyl trifluormethanesulfonate |
| MOMCl | chloromethyl methyl ether |
| NMO | N-methylmorpholine-N-Oxide |
| Proton sponge | 1,8-bis(dimethylamino)naphthalene |
| TBDPSCl | tert-butyldiphenylsilyl chloride |
| TBAF | tetrabutylammonium fluoride |
| TBSCl | tert-butyldimethylsilyl chloride |
| TBSOTf | tert-butyldimethylsilyl trifluoromethanesulfonate |
| Tebbe reagent | Bis(cyclopentadienyl)-μ-chloro-(dimethylaluminum)-μ-methylenetitanium |
| TEMPO | 2,2,6,6,-tetramethyl-1-piperidinyloxy |
| TPAP | tetrapropylammonium perruthenate |
| p-TSA | p-toluenesulfonic acid |
| White catalyst | 1,2-bis(phenylsulfinyl)ethane palladium (II) acetate |
| WHA | wound-healing assay |
| 4T1 | (mammary mouse cancer) |
| A549 | (lung carcinoma) |
| CMT-W1 | (canine mammary cancer) |
| CMT-W2 | (canine mammary cancer) |
| CMT-W1M | (canine lung metastasis) |
| CMT-W2M | (canine lung metastasis) |
| EC17 | (mouse esophageal cancer) |
| H1299 | (lung cancer) |
| H1975 | (lung adenocarcinoma) |
| HPAC | (human pancreas adenocarcinoma) |
| HUVECs | (human healthy endothelial cells) |
| LM2-4175 | (lung metastatic cells derived from MDA-MB-231) |
| Lovo | (human colon cancer) |
| MCF7 | (human breast cancer) |
| MDAB-MB-361 | (human breast cancer) |
| MCF-10A normal human mammary-gland epithelial | |
| MDA-MB-231 | (human breast cancer) |
| MDA-MB-435 | (human breast cancer) |
| P388/VCR | (vincristine-resistant mouse leukemia) |
| PC-3 | (human prostate cancer) |
| PDAC | (pancreatic ductal adenocarcinoma) |
| VJ-300 | (vincristine-resistant human epidermoid carcinoma) |
References
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Zhu, S. Small-molecule inhibitors of actin dynamics and cell motility. Curr. Top. Med. Chem. 2003, 3, 593–616. [Google Scholar] [CrossRef] [PubMed]
- Nakae, K.; Yoshimoto, Y.; Sawa, T.; Homma, Y.; Hamada, M.; Takeuchi, T.; Imoto, M. Migrastatin, a new inhibitor of tumor cell migration from Streptomyces sp. MK929-43F1. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 2000, 53, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Nakae, K.; Yoshimoto, Y.; Ueda, M.; Sawa, T.; Takahashi, Y.; Naganawa, H.; Takeuchi, T.; Imoto, M. Migrastatin, a novel 14-membered lactone from Streptomyces sp. MK929-43F1. J. Antibiot. 2000, 53, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.J.; Starks, C.M.; Carney, J.R.; Arslanian, R.; Cadapan, L.; Zavala, S.; Licari, P. Migrastatin and a new compound, isomigrastatin, from Streptomyces platensis. J. Antibiot. 2002, 55, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y.; Tashiro, E.; Imoto, M. Suppression of Multidrug Resistance by Migrastatin. J. Antibiot. 2006, 59, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Nakae, K.; Nishimura, Y.; Ohba, S.; Akamatsu, Y. Migrastatin Acts as a Muscarinic Acetylcholine Receptor Antagonist. J. Antibiot. 2006, 59, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Gaul, C.; Njardarson, J.T.; Danishefsky, S.J. The Total Synthesis of (+)-Migrastatin. J. Am. Chem. Soc. 2003, 125, 6042–6043. [Google Scholar] [CrossRef] [PubMed]
- Gaul, C.; Njardarson, J.T.; Shan, D.; Dorn, D.C.; Wu, K.-D.; Tong, W.P.; Huang, X.-Y.; Moore, M.A.S.; Danishefsky, S.J. The Migrastatin Family: Discovery of Potent Cell Migration Inhibitors by Chemical Synthesis. J. Am. Chem. Soc. 2004, 126, 11326–11337. [Google Scholar] [CrossRef] [PubMed]
- Reymond, S.; Cossy, J. Total Synthesis of (+)-Migrastatin. Eur. J. Org. Chem. 2006, 2006, 4800–4804. [Google Scholar] [CrossRef]
- Sai Baba, V.; Das, P.; Mukkanti, K.; Iqbal, J. A convergent synthesis of the macrolide core of migrastatin. Tetrahedron Lett. 2006, 47, 6083–6086. [Google Scholar] [CrossRef]
- Dias, L.C.; Finelli, F.G.; Conegero, L.S.; Krogh, R.; Andricopulo, A.D. Synthesis of the Macrolactone of Migrastatin and Analogues with Potent Cell-Migration Inhibitory Activity. Eur. J. Org. Chem. 2010, 2010, 6748–6759. [Google Scholar] [CrossRef] [Green Version]
- Gade, N.R.; Iqbal, J. Stereoselective formal synthesis of macrolidecore of migrastatin using late stage C–H oxidation. Tetrahedron Lett. 2013, 54, 4225–4227. [Google Scholar] [CrossRef]
- Lo Re, D.; Zhou, Y.; Nobis, M.; Anderson, K.I.; Murphy, P.V. Synthesis of migrastatin and its macroketone analogue and in vivo FRAP analysis of the macroketone on E-cadherin dynamics. Chembiochem. Eur. J. Chem. Biol. 2014, 15, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Reymond, S.; Cossy, J. Migrastatin and analogues: New anti-metastatic agents. C. R. Chim. 2008, 11, 1447–1462. [Google Scholar] [CrossRef]
- Gaul, C.; Danishefsky, S.J. Synthesis of the macrolide core of migrastatin. Tetrahedron Lett. 2002, 43, 9039–9042. [Google Scholar] [CrossRef]
- Reymond, S.; Cossy, J. Synthesis of migrastatin and its macrolide core. Tetrahedron 2007, 63, 5918–5929. [Google Scholar] [CrossRef]
- LoRe, D.; Zhou, Y.; Mucha, J.; Jones, L.F.; Leahy, L.; Santocanale, C.; Krol, M.; Murphy, P.V. Synthesis of Migrastatin Analogues as Inhibitors of Tumour Cell Migration: Exploring Structural Change in and on the Macrocyclic Ring. Chem. A Eur. J. 2015, 21, 18109–18121. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak, K.; Lo Re, D.; Gajewska, M.; Bulkowska, M.; Homa, A.; Pawlowski, K.; Motyl, T.; Murphy, P.V.; Krol, M. Migrastatin analogues inhibit canine mammary cancer cell migration and invasion. PLoS ONE 2013, 8, e76789. [Google Scholar] [CrossRef] [PubMed]
- Kitaori, K.; Furukawa, Y.; Yoshimoto, H.; Otera, J. CsF in organic synthesis. Regioselective nucleophilic reactions of phenols with oxiranes leading to enantiopure β-blockers. Tetrahedron 1999, 55, 14381–14390. [Google Scholar] [CrossRef]
- Xiang, G.; McLaughlin, L.W. A cytosine analogue containing a conformationally flexible acyclic linker for triplex formation at sites with contiguous G-C base pairs. Tetrahedron 1998, 54, 375–392. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Pearson, W.H.; Harvey, D.F.; Maring, C.J.; Springer, J.P. Chelation-controlled facially selective cyclocondensation reactions of chiral alkoxy aldehydes: Syntheses of a mouse androgen and of a carbon-linked disaccharide. J. Am. Chem. Soc. 1985, 107, 1256–1268. [Google Scholar] [CrossRef]
- Luche, J.L.; Gemal, A.L. Lanthanoids in organic synthesis. 5. Selective reductions of ketones in the presence of aldehydes. J. Am. Chem. Soc. 1979, 101, 5848–5849. [Google Scholar] [CrossRef]
- Ferrier, R.J. Unsaturated carbohydrates. Part II. Three reactions leading to unsaturated glycopyranosides. J. Chem. Soc. (Resum.) 1964, 5443–5449. [Google Scholar] [CrossRef]
- Tebbe, F.N.; Parshall, G.W.; Reddy, G.S. Olefin homologation with titanium methylene compounds. J. Am. Chem. Soc. 1978, 100, 3611–3613. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Organ. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Jørgensen, M.; Iversen, E.H.; Paulsen, A.L.; Madsen, R. Efficient Synthesis of Enantiopure Conduritols by Ring-Closing Metathesis. J. Organ. Chem. 2001, 66, 4630–4634. [Google Scholar] [CrossRef]
- Lee, W.-W.; Chang, S. A new and concise synthetic route to an enantiopure (+)-conduritol-E derivative from diethyl l-tartrate. Tetrahedron Asymm. 1999, 10, 4473–4475. [Google Scholar] [CrossRef]
- Njardarson, J.T.; Gaul, C.; Shan, D.; Huang, X.-Y.; Danishefsky, S.J. Discovery of Potent Cell Migration Inhibitors through Total Synthesis: Lessons from Structure–Activity Studies of (+)-Migrastatin. J. Am. Chem. Soc. 2004, 126, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Li, G.; Byun, H.-S.; Bittman, R. Synthesis of New Trans Double-Bond Sphingolipid Analogues: Δ4,6 and Δ6 Ceramides. J. Org. Chem. 2002, 67, 2600–2605. [Google Scholar] [CrossRef] [PubMed]
- Keck, G.E.; Savin, K.A.; Cressman, E.N.K.; Abbott, D.E. Effects of Olefin Geometry on the Stereochemistry of Lewis Acid Mediated Additions of Crotylstannanes to Aldehydes. J. Org. Chem. 1994, 59, 7889–7896. [Google Scholar] [CrossRef]
- Still, W.C.; Gennari, C. Direct synthesis of Z-unsaturated esters. A useful modification of the horner-emmons olefination. Tetrahedron Lett. 1983, 24, 4405–4408. [Google Scholar] [CrossRef]
- Patois, C.; Savignac, P.; About-Jaudet, E.; Collignon, N. A Convenient Method for the Synthesis of bis-Trifluoroethyl Phosphonoacetates. Synth. Commun. 1991, 21, 2391–2396. [Google Scholar] [CrossRef]
- Evans, D.A.; Gage, J.R.; Leighton, J.L. Total synthesis of (+)-calyculin A. J. Am. Chem. Soc. 1992, 114, 9434–9453. [Google Scholar] [CrossRef]
- Anderson, J.C.; McDermott, B.P.; Griffin, E.J. Asymmetric Epoxide Cyclisation Route to the F-pyran Fragment of the Altohyrtins and Key Aldol Studies. Tetrahedron 2000, 56, 8747–8767. [Google Scholar] [CrossRef]
- Yu, W.; Mei, Y.; Kang, Y.; Hua, Z.; Jin, Z. Improved Procedure for the Oxidative Cleavage of Olefins by OsO4–NaIO4. Org. Lett. 2004, 6, 3217–3219. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.D.; Kocienski, P.J.; Street, S.D.A. A Synthesis of (+)-Herboxidiene A. Synthesis 1996, 1996, 652–666. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Ando, K.; Oishi, T.; Hirama, M.; Ohno, H.; Ibuka, T. Z-Selective Horner–Wadsworth–Emmons Reaction of Ethyl (Diarylphosphono)acetates Using Sodium Iodide and DBU. J. Org. Chem. 2000, 65, 4745–4749. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Bartroli, J.; Shih, T.L. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar] [CrossRef]
- Evans, D.A.; Taber, T.R. Enantioselective aldol condensations via boron enolates. A steric model for asymmetric induction. Tetrahedron Lett. 1980, 21, 4675–4678. [Google Scholar] [CrossRef]
- Crimmins, M.T.; She, J. An Improved Procedure for Asymmetric Aldol Additions with N-Acyl Oxazolidinones, Oxazolidinethiones and Thiazolidinethiones. Synlett 2004, 2004, 1371–1374. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Tabet, E.A.; Chaudhary, K. Asymmetric Aldol Additions: Use of Titanium Tetrachloride and (−)-Sparteine for the Soft Enolization of N-Acyl Oxazolidinones, Oxazolidinethiones, and Thiazolidinethiones. J. Org. Chem. 2001, 66, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; Meira, P.R.R. Synthesis of C13-C22 fragment of the marine sponge polyketide callystatin A. Tetrahedron Lett. 2002, 43, 185–187. [Google Scholar] [CrossRef]
- Dias, L.C.; de Oliveira, L.G.; de Sousa, M.A. Total Synthesis of (−)-Pironetin. Org. Lett. 2003, 5, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; de Oliveira, L.G.; Vilcachagua, J.D.; Nigsch, F. Total Synthesis of (+)-Crocacin D. J. Org. Chem. 2005, 70, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; de Oliveira, L.G. Total Synthesis of (+)-Crocacin C. Org. Lett. 2001, 3, 3951–3954. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; de Oliveira, L.G. Short Synthesis of the 6,6-Spiroketal Cores of Spirofungins A and B. Org. Lett. 2004, 6, 2587–2590. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; de Castro, I.B.D.; Steil, L.J.; Augusto, T. A short approach to trisubstituted γ-butyrolactones. Tetrahedron Lett. 2006, 47, 213–216. [Google Scholar] [CrossRef]
- Evans, D.A.; Ratz, A.M.; Huff, B.E.; Sheppard, G.S. Total Synthesis of the Polyether Antibiotic Lonomycin A (Emericid). J. Am. Chem. Soc. 1995, 117, 3448–3467. [Google Scholar] [CrossRef]
- Ley, S.V.; Norman, J.; Griffith, W.P.; Marsden, S.P. Tetrapropylammonium Perruthenate, Pr4N+RuO4-, TPAP: A Catalytic Oxidant for Organic Synthesis. Synthesis 1994, 1994, 639–666. [Google Scholar] [CrossRef]
- Petasis, N.A.; Bzowej, E.I. Titanium-mediated carbonyl olefinations. 1. Methylenations of carbonyl compounds with dimethyltitanocene. J. Am. Chem. Soc. 1990, 112, 6392–6394. [Google Scholar] [CrossRef]
- Ando, K. Practical synthesis of Z-unsaturated esters by using a new Horner-Emmons reagent, ethyl diphenylphosphonoacetate. Tetrahedron Lett. 1995, 36, 4105–4108. [Google Scholar] [CrossRef]
- Ando, K. Highly Selective Synthesis of Z-Unsaturated Esters by Using New Horner–Emmons Reagents, Ethyl (Diarylphosphono)acetates. J. Org. Chem. 1997, 62, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Dias, L.C.; Ferreira, E.L. Allyltrichlorostannane additions to chiral dipeptide aldehydes. Tetrahedron Lett. 2001, 42, 7159–7162. [Google Scholar] [CrossRef]
- Dias, L.C.; Ferreira, A.A.; Diaz, G. Short Total Synthesis of Aspartyl Protease Inhibitors L-685,434, L-682,679 and L-685,458. Synlett 2002, 2002, 1845–1849. [Google Scholar] [CrossRef]
- Dias, L.C.; Diaz, G.; Ferreira, A.A.; Meira, P.R.R.; Ferreira, E. Allyltrichlorostannane Additions to α-Amino Aldehydes: Application to the Total Synthesis of the Aspartyl Protease Inhibitors L-682,679, L-684,414, L-685,434, and L-685,458. Synthesis 2003, 2003, 0603–0622. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Katz, J.D.; McAtee, L.C.; Tabet, E.A.; Kirincich, S.J. An Aldol Approach to the Synthesis of the EF Fragment of Spongistatin 1. Org. Lett. 2001, 3, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Ando, K. Z-Selective Horner–Wadsworth–Emmons Reaction of α-Substituted Ethyl (Diarylphosphono)acetates with Aldehydes1. J. Org. Chem. 1998, 63, 8411–8416. [Google Scholar] [CrossRef]
- Chen, M.S.; Prabagaran, N.; Labenz, N.A.; White, M.C. Serial Ligand Catalysis: A Highly Selective Allylic C−H Oxidation. J. Am. Chem. Soc. 2005, 127, 6970–6971. [Google Scholar] [CrossRef] [PubMed]
- Gormisky, P.E.; White, M.C. Synthetic Versatility in C–H Oxidation: A Rapid Approach to Differentiated Diols and Pyrans from Simple Olefins. J. Am. Chem. Soc. 2011, 133, 12584–12589. [Google Scholar] [CrossRef] [PubMed]
- Fraunhoffer, K.J.; Prabagaran, N.; Sirois, L.E.; White, M.C. Macrolactonization via Hydrocarbon Oxidation. J. Am. Chem. Soc. 2006, 128, 9032–9033. [Google Scholar] [CrossRef] [PubMed]
- Stang, E.M.; White, M.C. Total synthesis and study of 6-deoxyerythronolide B by late-stage C-H oxidation. Nat. Chem. 2009, 1, 547–551. [Google Scholar] [CrossRef]
- Stang, E.M.; White, M.C. On the Macrocyclization of the Erythromycin Core: Preorganization is Not Required. Angew. Chem. Int. Ed. 2011, 50, 2094–2097. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.C.; Jadhav, P.K.; Bhat, K.S. Chiral synthesis via organoboranes. 13. A highly diastereoselective and enantioselective addition of [(Z)-.gamma.-alkoxyallyl]diisopinocampheylboranes to aldehydes. J. Am. Chem. Soc. 1988, 110, 1535–1538. [Google Scholar] [CrossRef]
- Trost, B.M.; Malhotra, S.; Mino, T.; Rajapaksa, N.S. Dinuclear Zinc-Catalyzed Asymmetric Desymmetrization of Acyclic 2-Substituted-1,3-Propanediols: A Powerful Entry into Chiral Building Blocks. Chem. A Eur. J. 2008, 14, 7648–7657. [Google Scholar] [CrossRef] [PubMed]
- Maddess, M.L.; Tackett, M.N.; Watanabe, H.; Brennan, P.E.; Spilling, C.D.; Scott, J.S.; Osborn, D.P.; Ley, S.V. Total Synthesis of Rapamycin. Angew. Chem. Int. Ed. 2007, 46, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Li, X.; Nagorny, P.; Hayashida, J.; Danishefsky, S.J. A simple method for the conversion of carboxylic acids into thioacids with Lawesson’s reagent. Tetrahedron Lett. 2009, 50, 6684–6686. [Google Scholar] [CrossRef] [PubMed]
- Shipkova, M.; Wieland, E. Glucuronidation in therapeutic drug monitoring. Clin. Chim. Acta Int. J. Clin. Chem. 2005, 358, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, G.J.; Smith, T.W. Morphine-6-glucuronide: Actions and mechanisms. Med. Res. Rev. 2005, 25, 521–544. [Google Scholar] [CrossRef] [PubMed]
- Van Heek, M.; Farley, C.; Compton, D.S.; Hoos, L.; Alton, K.B.; Sybertz, E.J.; Davis, H.R. Comparison of the activity and disposition of the novel cholesterol absorption inhibitor, SCH58235, and its glucuronide, SCH60663. Br. J. Pharmacol. 2000, 129, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Stachulski, A.V.; Meng, X. Glucuronides from metabolites to medicines: A survey of the in vivo generation, chemical synthesis and properties of glucuronides. Nat. Prod. Rep. 2013, 30, 806–848. [Google Scholar] [CrossRef] [PubMed]
- El Alaoui, A.; Schmidt, F.; Monneret, C.; Florent, J.-C. Protecting Groups for Glucuronic Acid: Application to the Synthesis of New Paclitaxel (Taxol) Derivatives. J. Org. Chem. 2006, 71, 9628–9636. [Google Scholar] [CrossRef] [PubMed]
- Shimawaki, K.; Fujisawa, Y.; Sato, F.; Fujitani, N.; Kurogochi, M.; Hoshi, H.; Hinou, H.; Nishimura, S.-I. Highly Efficient and Versatile Synthesis of Proteoglycan Core Structures from 1,6-Anhydro-β-lactose as a Key Starting Material. Angew. Chem. Int. Ed. 2007, 46, 3074–3079. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, W.; Murphy, P.V. SnCl4- and TiCl4-Catalyzed Anomerization of Acylated O- and S-Glycosides: Analysis of Factors That Lead to Higher α:β Anomer Ratios and Reaction Rates. J. Org. Chem. 2010, 75, 6747–6755. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Poláková, M.; Pitt, N.; Tosin, M.; Murphy, P.V. Glycosidation–Anomerisation Reactions of 6,1-Anhydroglucopyranuronic Acid and Anomerisation of β-d-Glucopyranosiduronic Acids Promoted by SnCl4. Chem. A Eur. J. 2007, 13, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, W.; Murphy, P.V. α-Glycosphingolipids via Chelation-Induced Anomerization of O- and S-Glucuronic and Galacturonic Acid Derivatives. Org. Lett. 2009, 11, 939–942. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, C.; Murphy, P.V. Synthesis of α-S-Glycosphingolipids Based on Uronic Acids. Org. Lett. 2011, 13, 5168–5171. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.; Zhou, J.; Murphy, P.V. Regiospecific Anomerisation of Acylated Glycosyl Azides and Benzoylated Disaccharides by Using TiCl4. Chem. A Eur. J. 2013, 19, 14836–14851. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.W.; Mahon, M.F.; Murphy, P.V. Lewis Acid Induced Anomerization of Se-Glycosides. Application to Synthesis of α-Se-GalCer. Org. Lett. 2016, 18, 552–555. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.W.; Murphy, P.V. Synthesis of α-galactosyl ceramide analogues with an α-triazole at the anomeric carbon. Tetrahedron 2014, 70, 3191–3196. [Google Scholar] [CrossRef]
- Takemoto, Y.; Nakae, K.; Kawatani, M.; Takahashi, Y.; Naganawa, H.; Imoto, M. Migrastatin, a novel 14-membered ring macrolide, inhibits anchorage-independent growth of human small cell lung carcinoma Ms-1 cells. J. Antibiot. 2001, 54, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Kikuchi, J.; Sato, S.; Takano, H.; Saburi, Y.; Asoh, K.; Kuwano, M. Vincristine-resistant human cancer KB cell line and increased expression of multidrug-resistance gene. Jpn. J. Cancer Res. 1988, 79, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar] [PubMed]
- Shan, D.; Chen, L.; Njardarson, J.T.; Gaul, C.; Ma, X.; Danishefsky, S.J.; Huang, X.Y. Synthetic analogues of migrastatin that inhibit mammary tumor metastasis in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3772–3776. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Nagorny, P.; Krauss, I.J.; Perez, L.; Mandal, M.; Yang, G.; Ouerfelli, O.; Xiao, D.; Moore, M.A.S.; Massagué, J.; et al. Diverted Total Synthesis Leads to the Generation of Promising Cell-Migration Inhibitors for Treatment of Tumor Metastasis: In vivo and Mechanistic Studies on the Migrastatin Core Ether Analog. J. Am. Chem. Soc. 2010, 132, 3224–3228. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, N.; Njardarson, J.T.; Nagorny, P.; Yang, G.; Downey, R.; Ouerfelli, O.; Moore, M.A.; Danishefsky, S.J. Emergence of potent inhibitors of metastasis in lung cancer via syntheses based on migrastatin. Proc. Natl. Acad. Sci. USA 2011, 108, 15074–15078. [Google Scholar] [CrossRef] [PubMed]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Reduction of Established Spontaneous Mammary Carcinoma Metastases following Immunotherapy with Major Histocompatibility Complex Class II and B7.1 Cell-based Tumor Vaccines. Cancer Res. 1998, 58, 1486–1493. [Google Scholar] [PubMed]
- Aslakson, C.J.; Miller, F.R. Selective Events in the Metastatic Process Defined by Analysis of the Sequential Dissemination of Subpopulations of a Mouse Mammary Tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar] [PubMed]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massagué, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, S.; Jakoncic, J.; Zhang, J.J.; Huang, X.Y. Migrastatin analogues target fascin to block tumour metastasis. Nature 2010, 464, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.J.; Tso, E.; Skacel, M.; Pettay, J.; Tarr, S.; Budd, T.; Tubbs, R.R.; Adams, J.C.; Hicks, D.G. The expression of fascin, an actin-bundling motility protein, correlates with hormone receptor-negative breast cancer and a more aggressive clinical course. Clin. Cancer Res. 2005, 11, 186–192. [Google Scholar] [PubMed]
- Grothey, A.; Hashizume, R.; Sahin, A.A.; McCrea, P.D. Fascin, an actin-bundling protein associated with cell motility, is upregulated in hormone receptor negative breast cancer. Br. J. Cancer 2000, 83, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Parsons, M.; Adams, J.C. Dual Actin-bundling and Protein Kinase C-binding Activities of Fascin Regulate Carcinoma Cell Migration Downstream of Rac and Contribute to Metastasis. Mol. Biol. Cell 2007, 18, 4591–4602. [Google Scholar] [CrossRef] [PubMed]
- Vignjevic, D.; Schoumacher, M.; Gavert, N.; Janssen, K.P.; Jih, G.; Lae, M.; Louvard, D.; Ben-Ze’ev, A.; Robine, S. Fascin, a novel target of beta-catenin-TCF signaling, is expressed at the invasive front of human colon cancer. Cancer Res. 2007, 67, 6844–6853. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro-Matsumura, S.; Matsumura, F. Purification and characterization of an F-actin-bundling 55-kilodalton protein from HeLa cells. J. Biol. Chem. 1985, 260, 5087–5097. [Google Scholar] [PubMed]
- Nagorny, P.; Krauss, I.; Njardarson, J.T.; Perez, L.; Gaul, C.; Yang, G.; Ouerfelli, O.; Danishefsky, S.J. Confirmation of the Structures of Synthetic Derivatives of Migrastatin in the Light of Recently Disclosed Crystallographically Based Claims. Tetrahedron Lett. 2010, 51, 3873–3875. [Google Scholar] [CrossRef] [PubMed]
- Krol, M.; Polanska, J.; Pawlowski, K.M.; Turowski, P.; Skierski, J.; Majewska, A.; Ugorski, M.; Morty, R.E.; Motyl, T. Transcriptomic signature of cell lines isolated from canine mammary adenocarcinoma metastases to lungs. J. Appl. Genet. 2010, 51, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak, K.; Pawłowski, K.M.; Orzechowska, E.J.; Dolka, I.; Mucha, J.; Motyl, T.; Król, M. A role of ghrelin in canine mammary carcinoma cells proliferation, apoptosis and migration. BMC Vet. Res. 2012, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Król, M.; Pawłowski, K.M.; Majchrzak, K.; Gajewska, M.; Majewska, A.; Motyl, T. Global gene expression profiles of canine macrophages and canine mam mary cancer cells grown as a co-culture in vitro. BMC Vet. Res. 2012, 8, 16. [Google Scholar]
- Manuali, E.; De Giuseppe, A.; Feliziani, F.; Forti, K.; Casciari, C.; Marchesi, M.C.; Pacifico, E.; Pawłowski, K.M.; Majchrzak, K.; Król, M. CA 15–3 cell lines and tissue expression in canine mammary cancer and the correlation between serum levels and tumour histological grade. BMC Vet. Res. 2012, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Vignjevic, D.; Kojima, S.; Aratyn, Y.; Danciu, O.; Svitkina, T.; Borisy, G.G. Role of fascin in filopodial protrusion. J. Cell Biol. 2006, 174, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Mejillano, M.R.; Kojima, S.; Applewhite, D.A.; Gertler, F.B.; Svitkina, T.M.; Borisy, G.G. Lamellipodial versus filopodial mode of the actin nanomachinery: Pivotal role of the filament barbed end. Cell 2004, 118, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G. Snail1 links transcriptional control with epigenetic regulation. EMBO J. 2010, 29, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Rajasekaran, A.K. Dishonorable Discharge: The Oncogenic Roles of Cleaved E-Cadherin Fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of Members of the Cadherin Superfamily in Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, W.; Behrens, J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta (BBA) Rev. Cancer 1994, 1198, 11–26. [Google Scholar] [CrossRef]
- Risinger, J.I.; Berchuck, A.; Kohler, M.F.; Boyd, J. Mutations of the E-cadherin gene in human gynecologic cancers. Nat. Genet. 1994, 7, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Cleton-Jansen, A.M.; Nollet, F.; de Leeuw, W.J.; van de Vijver, M.; Cornelisse, C.; van Roy, F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995, 14, 6107–6115. [Google Scholar] [PubMed]
- Efstathiou, J.A.; Liu, D.; Wheeler, J.M.D.; Kim, H.C.; Beck, N.E.; Ilyas, M.; Karayiannakis, A.J.; Mortensen, N.J.M.; Kmiot, W.; Playford, R.J.; et al. Mutated epithelial cadherin is associated with increased tumorigenicity and loss of adhesion and of responsiveness to the motogenic trefoil factor 2 in colon carcinoma cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Authors | n0 Steps | Overal Yield | Key Steps | Notes | Reference |
|---|---|---|---|---|---|---|
| 2004 | Danishefsky et al. | 10 | 22% | LACDAC reaction, Ferrier rearrangement | Multi-gram scale synthesis | [8,9,16] |
| 2006 | Cossy et al. | 11 (+4) a | 11% | Stereoselective crotylmetalation, RCM | - | [10] |
| 2007 | Cossy et al. | 11 (+4) a | 35% | Stereoselective crotylmetalation, Still-Gennari olefination | Entry to isomigrastatin analogues, gram scale | [17] |
| 2006 | Lqbal et al. | 12 | 8% | Evans aldol condensation and distereoselective vinylmagnesium bromide additon | Entry to isomigrastatin analogues | [11] |
| 2010 | Dias et al. | 14 | 1.2% | Upjohn dihydroxilation, Horner-Wadsworth-Emmons olefination | Entry to isomigrastatin-core epimers, gram scale | [12] |
| 2013 | Lqbal et al. | 11 | 5.9% | Pd(II) catalyzed intramolecular C-H oxidation | Entry to migrastatin-core with different functional groups | [13] |
| 2014 | Murphy et al. | 9 | 30% | Brown alcoxyallylation, HWE, Zinc catalyzed asimmetric desymmetrization | Entry to isomigrastatin analogues, gram scale | [14,18,19] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giralt, E.; Lo Re, D. The Therapeutic Potential of Migrastatin-Core Analogs for the Treatment of Metastatic Cancer. Molecules 2017, 22, 198. https://doi.org/10.3390/molecules22020198
Giralt E, Lo Re D. The Therapeutic Potential of Migrastatin-Core Analogs for the Treatment of Metastatic Cancer. Molecules. 2017; 22(2):198. https://doi.org/10.3390/molecules22020198
Chicago/Turabian StyleGiralt, Ernest, and Daniele Lo Re. 2017. "The Therapeutic Potential of Migrastatin-Core Analogs for the Treatment of Metastatic Cancer" Molecules 22, no. 2: 198. https://doi.org/10.3390/molecules22020198
APA StyleGiralt, E., & Lo Re, D. (2017). The Therapeutic Potential of Migrastatin-Core Analogs for the Treatment of Metastatic Cancer. Molecules, 22(2), 198. https://doi.org/10.3390/molecules22020198