
Synthesis, X-ray Single Crystal Structure, Molecular Docking and DFT Computations on N-[(1E)-1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A New Potential Antifungal Agent Precursor

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
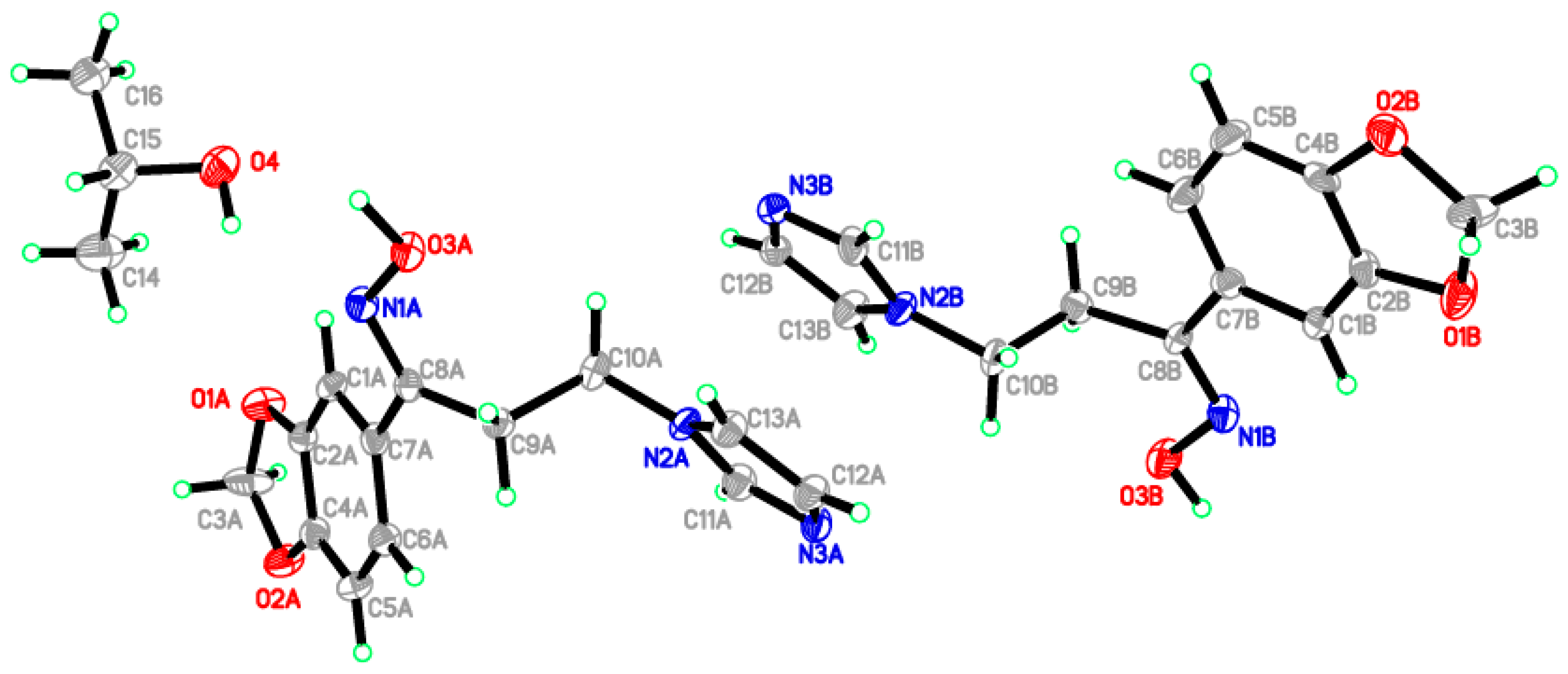
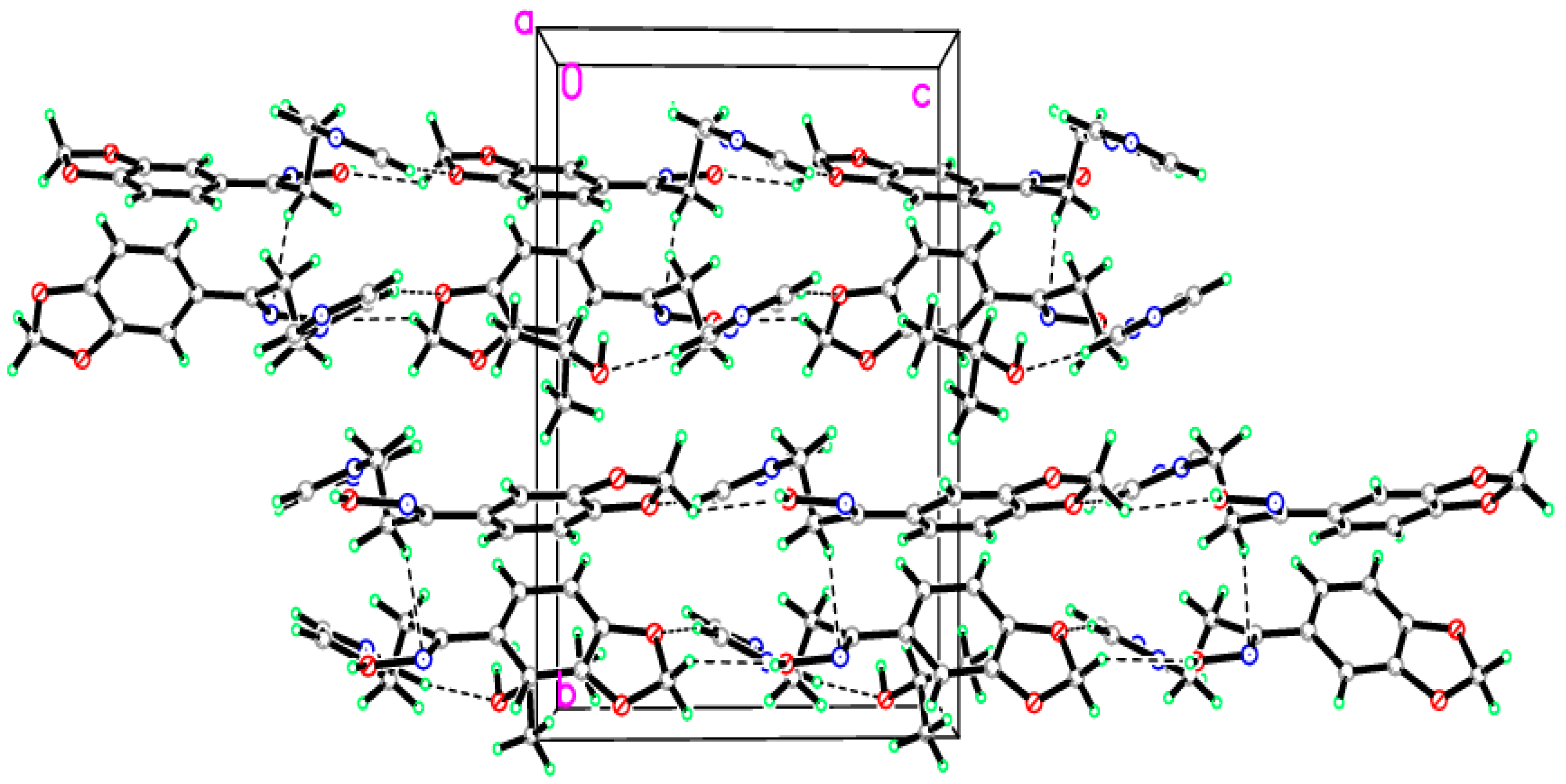
2.2. Crystal Structure of the Title Compound 4
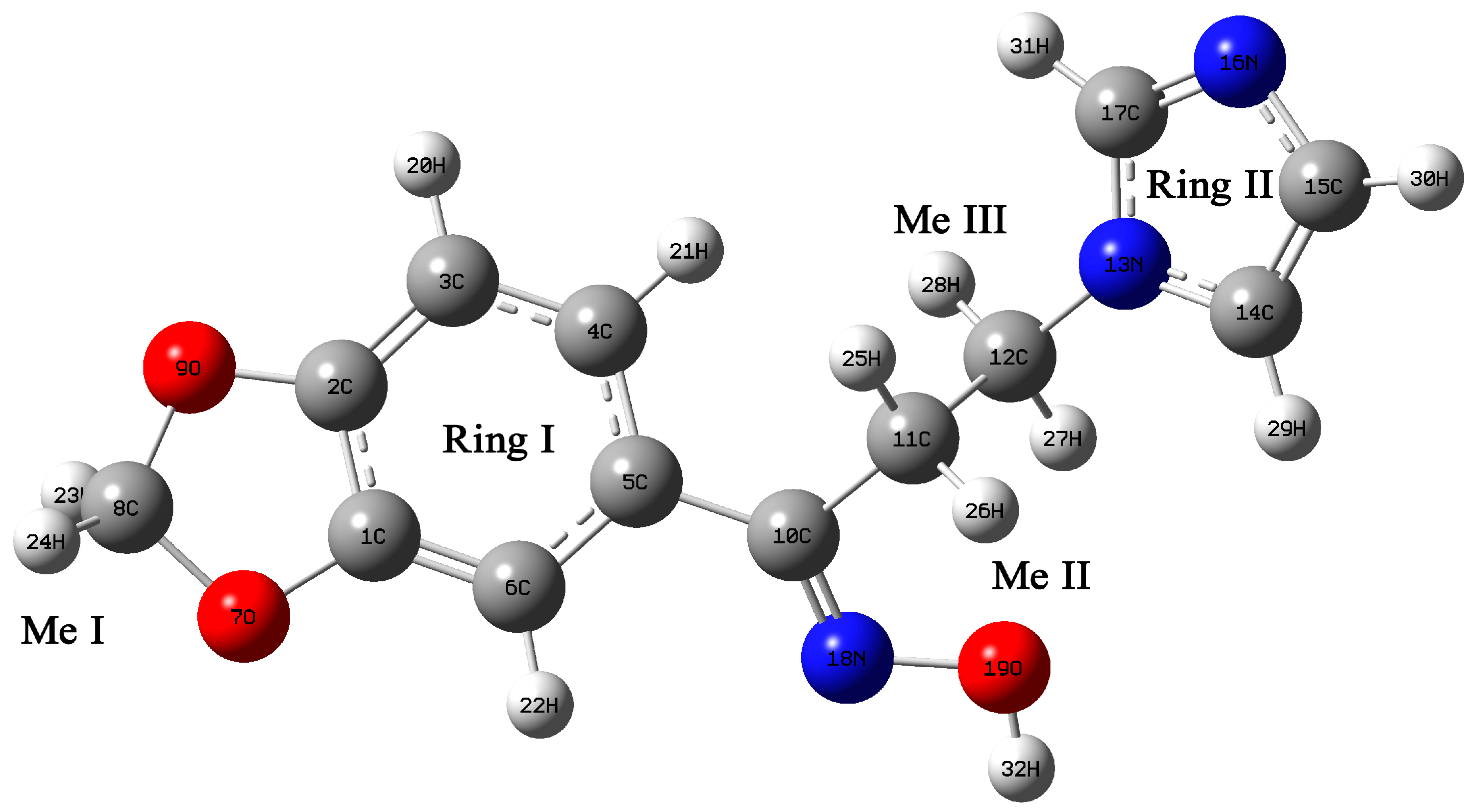
2.3. Structural Geometry Analysis
2.4. Natural Bond Orbital Analysis
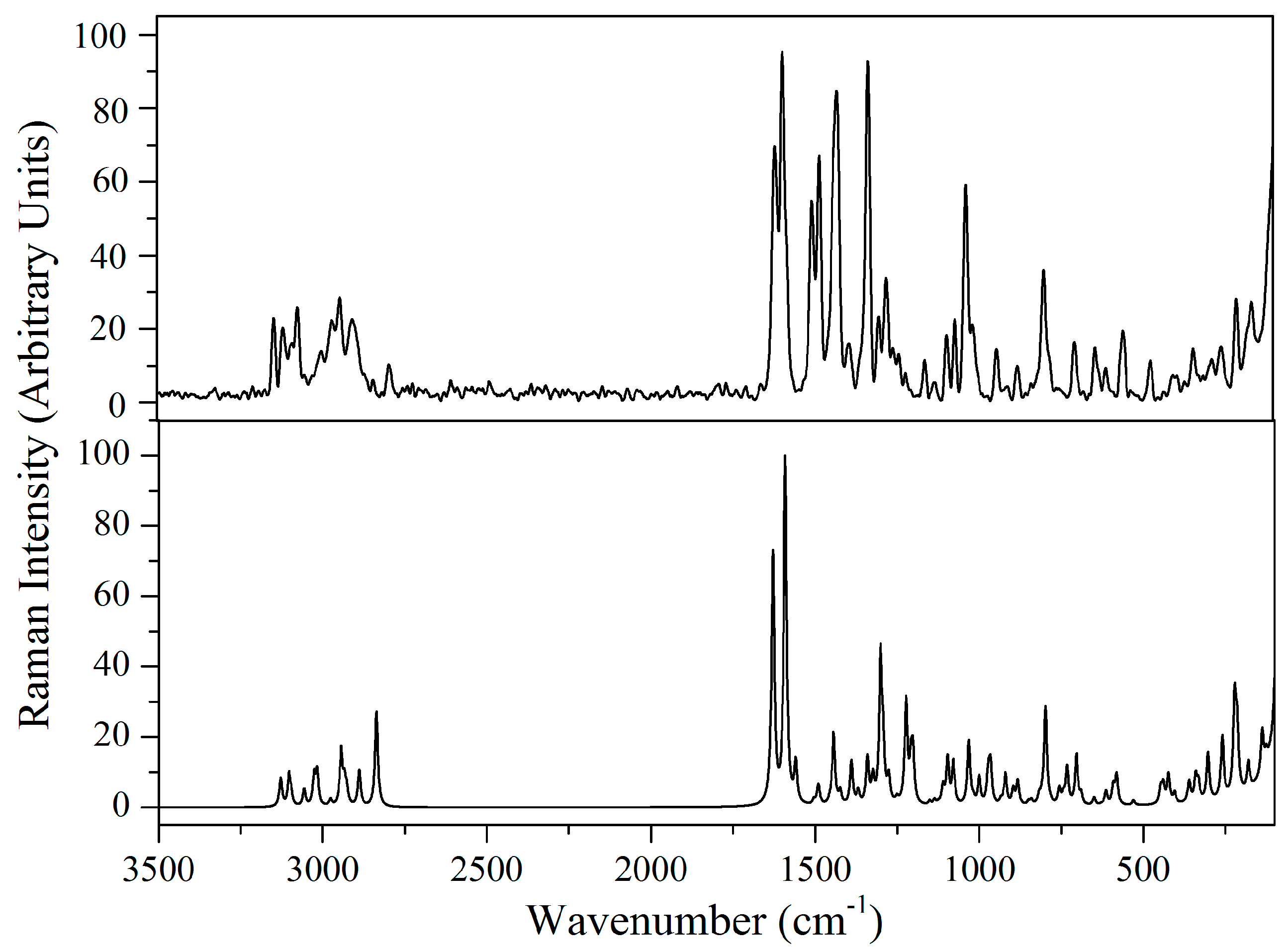
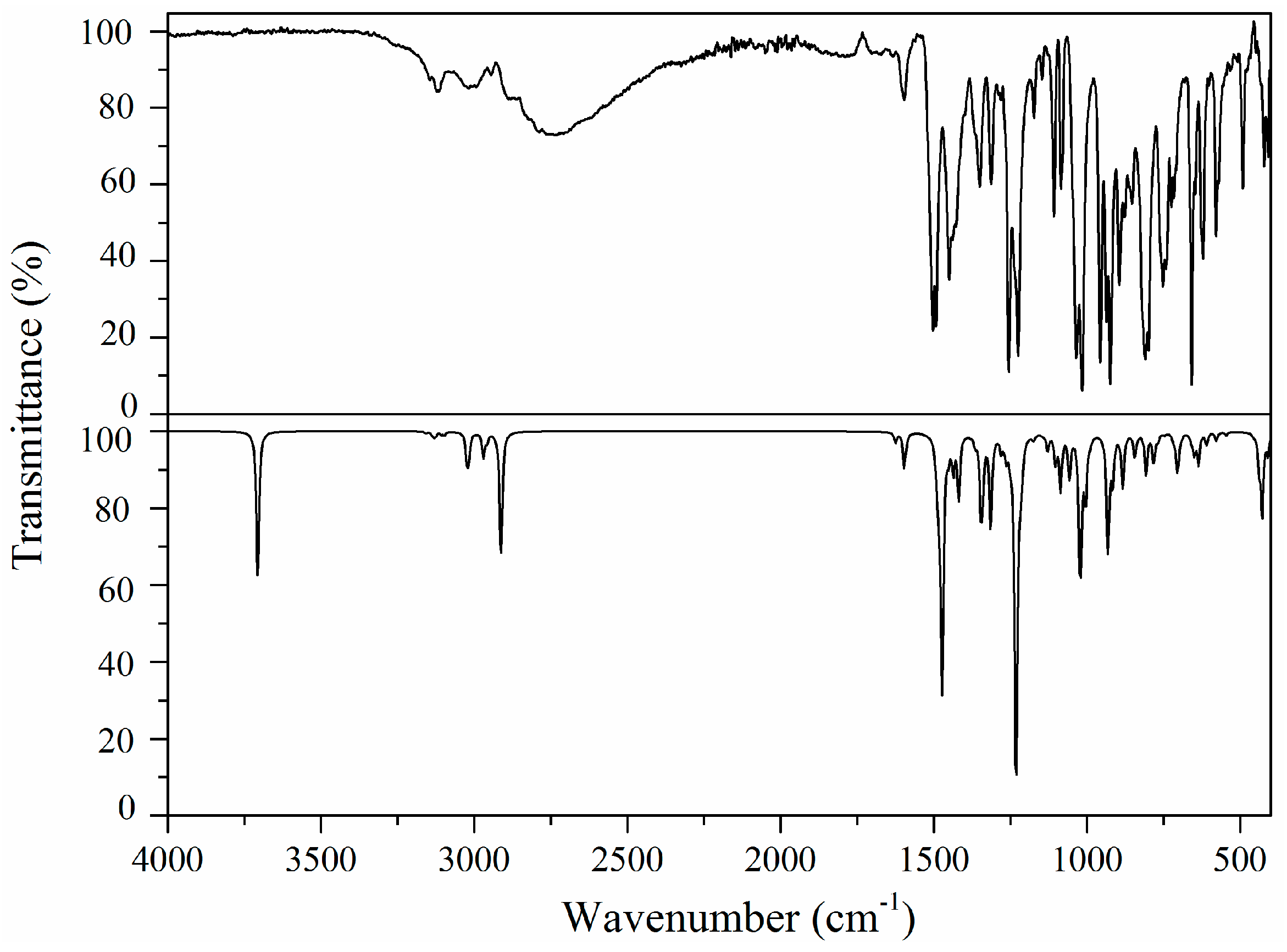
2.5. Vibrational Analysis
2.5.1. Imidazole Ring Vibrations
2.5.2. Methylene Group Vibrations
2.5.3. Benzodioxole Ring Vibrations
2.6. Frontier Molecular Orbital Analysis
2.7. NMR Chemical Shift Analysis
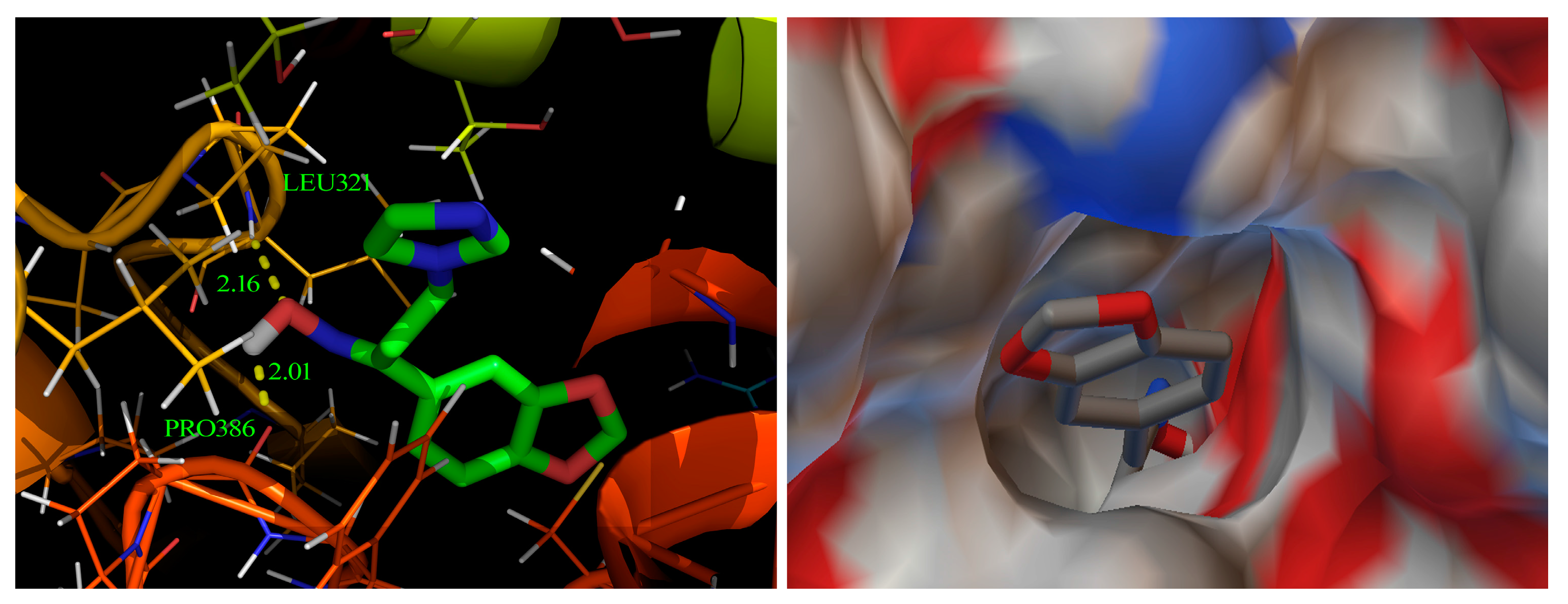
2.8. Molecular Docking Study
2.9. Antifungal Activity of the Title Compound 4
3. Experimental
3.1. General
3.2. Synthesis
3.2.1. Synthesis of 1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propan-1-one (3)
3.2.2. Synthesis of (1E)-1-(2H-1,3-Benzodioxol-5-yl)-N-hydroxy-3-(1H-imidazol-1-yl)propan-1-imine (4)
3.3. Crystal Structure Determination
3.4. FT-IR and FT-Raman Measurements
3.5. Quantum Chemical Calculations
3.6. Antifungal Activity
3.6.1. Materials
3.6.2. Organisms
3.6.3. Preparation of Fungal Inocula
3.6.4. Preparation of the Tested Compound Solution
3.6.5. Antifungal Susceptibility Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Caston-Osorio, J.J.; Rivero, A.; Torre-Cisneros, J. Epidemiology of invasive fungal infection. Int. J. Antimicrob. Agents 2008, 32 (Suppl. 2), S103–S109. [Google Scholar] [CrossRef]
- Cuenca-Estrella, M.; Bernal-Martinez, L.; Buitrago, M.J.; Castelli, M.V.; Gomez-Lopez, A.; Zaragoza, O.; Rodriguez-Tudela, J.L. Update on the epidemiology and diagnosis of invasive fungal infection. Int. J. Antimicrob. Agents 2008, 32 (Suppl. 2), S143–S147. [Google Scholar] [CrossRef]
- Denning, D.W.; Kibbler, C.C.; Barnes, R.A. British Society for Medical Mycology proposed standards of care for patients with invasive fungal infections. Lancet Infect. Dis. 2003, 3, 230–240. [Google Scholar] [CrossRef]
- Kathiravan, M.K.; Salake, A.B.; Chothe, A.S.; Dudhe, P.B.; Watode, R.P.; Mukta, M.S.; Gadhwe, S. The biology and chemistry of antifungal agents: A review. Bioorg. Med. Chem. 2012, 20, 5678–5698. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Messer, S.A.; Hollis, R.J.; Jones, R.N. Antifungal activities of posaconazole, ravuconazole, and voriconazole compared to those of itraconazole and amphotericin B against 239 clinical isolates of Aspergillus spp. and other filamentous fungi: Report from SENTRY Antimicrobial Surveillance Program, 2000. Antimicrob. Agents Chemother. 2002, 46, 1032–1037. [Google Scholar] [PubMed]
- Spreghini, E.; Maida, C.M.; Tomassetti, S.; Orlando, F.; Giannini, D.; Milici, M.E.; Scalise, G.; Barchiesi, F. Posaconazole against Candida glabrata isolates with various susceptibilities to fluconazole. Antimicrob. Agents Chemother. 2008, 52, 1929–1933. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, Y.; Yoshida, Y.; Sato, R. Yeast cytochrome P-450 catalyzing lanosterol 14 α-demethylation. II. Lanosterol metabolism by purified P-450(14)DM and by intact microsomes. J. Biol. Chem. 1984, 259, 1661–1666. [Google Scholar] [PubMed]
- Vanden Bossche, H.; Koymans, L. Cytochromes P450 in fungi. Mycoses 1998, 41 (Suppl. 1), 32–38. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Enein, M.N.; El-Azzouny, A.A.; Attia, M.I.; Saleh, O.A.; Kansoh, A.L. Synthesis and anti-Candida potential of certain novel 1-[(3-substituted-3-phenyl)propyl]-1H-imidazoles. Arch. Pharm. 2011, 344, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Mares, M.; Nastasa, V. A novel antifungal agent with broad spectrum: 1-(4-biphenylyl)-3-(1H-imidazol-1-yl)-1-propanone. Arch. Pharm. 2013, 346, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.I.; Radwan, A.A.; Zakaria, A.S.; Almutairi, M.S.; Ghoneim, S.W. 1-Aryl-3-(1H-imidazol-1-yl)propan-1-ol esters: Synthesis, anti-Candida potential and molecular modeling studies. Chem. Cent. J. 2013, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cotinguiba, F.; Regasini, L.O.; da Silva Bolzani, V.S.; Debonsi, H.; Passerini, G.D.; Cicarelli, R.M.B.; Kato, M.J.; Furlan, M. Piperamides and their derivatives as potential antitrypanosomal agents. Med. Chem. Res. 2009, 18, 703–711. [Google Scholar] [CrossRef]
- Himaja, M.; Vandana, K.; Ranjitha, A.; Ramana, M.; Karigar, A. Synthesis, docking studies and antioxidant activity of 1, 3-benzodioxole-5-carboxyl amino acids and dipeptides. Int. Res. J. Pharm. 2011, 2, 57–61. [Google Scholar]
- Leite, A.C.; da Silva, K.P.; de Souza, I.A.; de Araujo, J.M.; Brondani, D.J. Synthesis, antitumour and antimicrobial activities of new peptidyl derivatives containing the 1,3-benzodioxole system. Eur. J. Med. Chem. 2004, 39, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Enein, M.N.; El-Azzouny, A.A.; Attia, M.I.; Maklad, Y.A.; Amin, K.M.; Abdel-Rehim, M.; El-Behairy, M.F. Design and synthesis of novel stiripentol analogues as potential anticonvulsants. Eur. J. Med. Chem. 2012, 47, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.I.; El-Brollosy, N.R.; Kansoh, A.L.; Ghabbour, H.A.; Al-Wabli, R.I.; Fun, H.-K. Synthesis, single crystal X-ray structure, and antimicrobial activity of 6-(1,3-benzodioxol-5-ylmethyl)-5-ethyl-2-{[2-(morpholin-4-yl)ethyl]sulfanyl}pyrimidin-4(3H)-one. J. Chem. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Fun, H.-K.; Quah, C.K.; Attia, M.I.; Almutairi, M.S.; Ghoneim, S.W. (E)-N-[3-(imidazol-1-yl)-1-phenyl-propylidene]hydroxylamine. Acta Cryst. E 2012, 68, o627. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Murakami, R.; Noda, I.; Ozaki, Y. Infrared and Raman spectroscopy and quantum chemistry calculation studies of C–H⋯O hydrogen bondings and thermal behavior of biodegradable polyhydroxyalkanoate. J. Mol. Struct. 2005, 744, 35–46. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic basis of improper hydrogen bonding: A subtle balance of hyperconjugation and rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef] [PubMed]
- Rauhut, G.; Pulay, P. Transferable scaling factors for density functional derived vibrational force fields. J. Phys. Chem. 1995, 99, 3093–3100. [Google Scholar] [CrossRef]
- Fogarasi, G.; Zhou, X.; Taylor, P.W.; Pulay, P. The calculation of ab initio molecular geometries: Efficient optimization by natural internal coordinates and empirical correction by offset forces. J. Am. Chem. Soc. 1992, 114, 8191–8201. [Google Scholar] [CrossRef]
- Billes, F.; Endrédi, H.; Jalsovszky, G. Vibrational spectroscopy of diazoles. J. Mol. Struct. THEOCHEM 1999, 465, 157–172. [Google Scholar] [CrossRef]
- Loeffen, P.W.; Pettifer, R.F.; Fillaux, F.; Kearley, G.J. Vibrational force field of solid imidazole from inelastic neutron scattering. J. Chem. Phys. 1995, 103, 8444–8455. [Google Scholar] [CrossRef]
- Naumov, P.; Ristova, M.; Šoptrajanov, B.; Zugik, M. Vibrational spectra of bis(acetato)tetrakis(imidazole)copper(II). J. Mol. Struct. 2001, 598, 235–243. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: New York, NY, USA, 1999. [Google Scholar]
- Mesu, J.G.; Visser, T.; Soulimani, F.; Weckhuysen, B.M. Infrared and Raman spectroscopic study of pH-induced structural changes of l-histidine in aqueous environment. Vib. Spectrosc. 2005, 39, 114–125. [Google Scholar] [CrossRef]
- Ditchfield, R. Molecular orbital theory of magnetic shielding and magnetic susceptibility. J. Chem. Phys. 1972, 56, 5688–5691. [Google Scholar] [CrossRef]
- Baxter, C.A.; Murray, C.W.; Waszkowycz, B.; Li, J.; Sykes, R.A.; Bone, R.G.; Perkins, T.D.; Wylie, W. New approach to molecular docking and its application to virtual screening of chemical databases. J. Chem. Inf. Comput. Sci. 2000, 40, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian-09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Podust, L.M.; Poulos, T.L.; Waterman, M.R. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Nat. Acad. Sci. USA 2001, 98, 3068–3073. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- APEX2, SAINT and SADABS; Brucker AXS: Madison, WI, USA, 2009.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sundius, T. Molvib-A flexible program for force field calculations. J. Mol. Struct. 1990, 218, 321–326. [Google Scholar] [CrossRef]
- Sundius, T. Scaling of ab initio force fields by MOLVIB. Vib. Spectrosc. 2002, 29, 89–95. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO, version 3.1; Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Rodriguez-Tudela, J.L.; Arendrup, M.C.; Barchiesi, F.; Bille, J.; Chryssanthou, E.; Cuenca-Estrella, M.; Dannaoui, E.; Denning, D.W.; Donnelly, J.P.; Dromer, F.; et al. EUCAST Definitive Document EDef 7.1: method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts: Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST). Clin. Microbiol. Infect. 2008, 14, 398–405. [Google Scholar]
- Sample Availability: Samples of the synthesized compounds are available from the corresponding author.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| O3A–H3OA···N3A i | 1.00(5) | 1.73(5) | 2.722(5) | 176(6) |
| O3B–H3OB···N3B ii | 0.98(6) | 1.70(6) | 2.672(5) | 175(8) |
| O4–H1O4···N1A | 0.91(7) | 2.38(7) | 3.127(6) | 139(6) |
| C3A–H3AB···O3A iii | 0.9900 | 2.4900 | 3.128(6) | 122.00 |
| C3B–H3BB···O3B iv | 0.9900 | 2.3600 | 3.080(6) | 129.00 |
| C11A–H11A···O4 ii | 0.9500 | 2.2700 | 3.179(7) | 159.00 |
| C13A–H13A···O2A iv | 0.9500 | 2.5600 | 3.459(6) | 158.00 |
| C13B–H13B···O2B iii | 0.9500 | 2.5200 | 3.424(6) | 160.00 |
| C9B–H9BB···N1A v | 0.9900 | 2.5800 | 3.502(6) | 154.00 |
| Bond Length (Å) | Bond Angle (°) | Dihedral Angle (°) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | Calculated | Exp. | Parameters | Calculated | Exp. | Parameters | Calculated | Exp. | |||
| Gas Phase | Solution Phase | Gas Phase | Solution Phase | Gas Phase | Solution Phase | ||||||
| C1–C2 | 1.3947 | 1.3945 | 1.379 | C1–C2–C3 | 121.40 | 121.44 | 121.60 | C1–C2–C3–C4 | –0.32 | –0.29 | 1.20 |
| C2–C3 | 1.3753 | 1.3764 | 1.362 | C1–C2–O9 | 109.69 | 109.67 | 110.05 | C1–C2–C3–H20 | 179.6 | 179.72 | –179.12 |
| C3–C4 | 1.4049 | 1.405 | 1.395 | C1–C6–C5 | 117.55 | 117.55 | 117.05 | C1–C2–O9–C8 | 10.37 | 10.84 | –4.10 |
| C4–C5 | 1.4001 | 1.4005 | 1.394 | C1–C6–H22 | 121.74 | 121.53 | 121.62 | C1–O7–C8–O9 | 16.91 | 17.41 | –4.55 |
| C5–C6 | 1.4181 | 1.4183 | 1.411 | C1–O7–C8 | 105.45 | 105.50 | 105.41 | C1–O7–C8–H23 | 135.54 | 135.89 | 115.78 |
| C6–C1 | 1.3707 | 1.3719 | 1.370 | C2–C1–C6 | 122.44 | 122.37 | 122.65 | C2–C1–O7–C8 | –10.64 | –10.95 | 1.95 |
| C1–O7 | 1.3743 | 1.374 | 1.376 | C2–C1–O7 | 109.26 | 109.35 | 109.85 | C2–C3–C4–C5 | 0.39 | 0.51 | –1.00 |
| C2–O9 | 1.3695 | 1.3695 | 1.382 | C2–C3–C4 | 116.93 | 116.98 | 117.25 | C2–C3–C4–H21 | –179.8 | –179.96 | 178.51 |
| C3–H20 | 1.0821 | 1.0822 | 0.950 | C2–O9–C8 | 105.31 | 105.44 | 105.57 | C2–C1–C6–C5 | 0.21 | 0.09 | 1.80 |
| C4–H21 | 1.0814 | 1.0815 | 0.950 | C3–C2–O9 | 128.89 | 128.87 | 128.35 | C2–C1–C6–H22 | –179.59 | 179.94 | –179.81 |
| C5–C10 | 1.4846 | 1.4847 | 1.482 | C2–C3–H20 | 121.55 | 121.72 | 121.41 | C3–C4–C5–C6 | –0.17 | –0.44 | 1.05 |
| C6–H22 | 1.0805 | 1.0808 | 0.950 | C4–C3–H20 | 121.52 | 121.30 | 121.44 | C3–C2–C1–O7 | –178.62 | –178.62 | 179.60 |
| C8–O7 | 1.4317 | 1.4372 | 1.424 | C3–C4–C5 | 122.28 | 122.18 | 121.95 | C3–C4–C5–C10 | 178.73 | 178.73 | 177.75 |
| C8–O9 | 1.4360 | 1.4401 | 1.418 | C3–C4–H21 | 117.57 | 117.55 | 119.03 | C3–C2–O9–C8 | –170.62 | –170.62 | 177.8 |
| C8–H23 | 1.0892 | 1.0877 | 0.990 | C5–C4–H21 | 120.15 | 120.27 | 119.03 | C4–C5–C6–C1 | 0.13 | 0.13 | 2.30 |
| C8–H24 | 1.0966 | 1.0943 | 0.990 | C4–C5–C6 | 119.38 | 119.48 | 119.62 | C4–C5–C6–H22 | –179.72 | –179.72 | 179.78 |
| C10–C11 | 1.5127 | 1.5133 | 1.519 | C4–C5–C10 | 121.01 | 120.74 | 121.35 | C4–C5–C10–C11 | 16.37 | 16.37 | 19.05 |
| C10–N18 | 1.2861 | 1.2868 | 1.288 | C5–C10–C11 | 121.63 | 121.26 | 119.86 | C4–C5–C10–N18 | –163.31 | –163.31 | 164.15 |
| C11–C12 | 1.5435 | 1.5427 | 1.523 | C5–C10–N18 | 116.24 | 116.28 | 117.56 | C6–C1–C2–C3 | 0.002 | 0.004 | –1.60 |
| C11–H25 | 1.0897 | 1.0894 | 0.990 | C5–C6–H22 | 120.71 | 120.91 | 121.63 | C6–C1–C2–O9 | 178.67 | 178.67 | 178.55 |
| C11–H26 | 1.0912 | 1.0915 | 0.990 | C6–C5–C10 | 119.60 | 119.78 | 118.95 | C6–C1–O7–C8 | 170.55 | 170.55 | –178.00 |
| C12–N13 | 1.4574 | 1.4620 | 1.459 | C6–C1–O7 | 128.27 | 128.27 | 127.61 | C6–C5–C4–H21 | –179.7 | –179.96 | –179.51 |
| C12–H27 | 1.0902 | 1.0894 | 0.990 | O7–C8–O9 | 107.29 | 106.84 | 109.45 | C6–C5–C10–C11 | –164.46 | –164.46 | 159.60 |
| C12–H28 | 1.0924 | 1.0911 | 0.990 | O7–C8–H23 | 109.53 | 109.56 | 109.83 | C6–C5–C10–N18 | 15.85 | 15.85 | 17.25 |
| N13–C14 | 1.3815 | 1.3802 | 1.370 | O7–C8–H24 | 109.50 | 109.44 | 109.81 | O7–C1–C2–O9 | 0.06 | 0.13 | 1.40 |
| N13–C17 | 1.3678 | 1.3629 | 1.349 | O9–C8–H23 | 109.38 | 109.46 | 109.80 | O7–C1–C6–C5 | 178.42 | 178.54 | 179.65 |
| C14–C15 | 1.3716 | 1.3713 | 1.360 | O9–C8–H24 | 109.17 | 109.18 | 109.81 | O7–C1–C6–H22 | –1.73 | –1.64 | 0.59 |
| C14–H29 | 1.0778 | 1.0776 | 0.950 | C10–C11–C12 | 111.71 | 111.99 | 111.31 | O9–C2–C3–C4 | –178.68 | –178.61 | 178.55 |
| C15–N16 | 1.3751 | 1.3795 | 1.377 | C10–C11–H25 | 110.75 | 110.32 | 109.42 | O9–C2–C3–H20 | 1.32 | 1.52 | –1.53 |
| C15–H30 | 1.0790 | 1.0792 | 0.950 | C10–C11–H26 | 108.46 | 108.41 | 109.42 | C10–C5–C6–C1 | –179.05 | –179.29 | –178.45 |
| N16–C17 | 1.3142 | 1.3197 | 1.323 | C11–C10–N18 | 122.12 | 122.46 | 122.83 | C10–C5–C4–H21 | –0.79 | –1.78 | –1.11 |
| C17–H31 | 1.0802 | 1.0798 | 0.950 | C11–C12–N13 | 112.49 | 112.05 | 111.10 | C10–C5–C6–H22 | 1.09 | 0.90 | 1.52 |
| N18–O19 | 1.4067 | 1.4061 | 1.409 | C12–C11–H25 | 109.89 | 109.89 | 109.42 | C5–C4–C3–H20 | –179.53 | –179.49 | 178.63 |
| O19–H32 | 0.9631 | 0.9647 | 0.985 | C12–C11–H26 | 108.47 | 108.56 | 109.43 | C12–N13–C14–C15 | 176.72 | 177.67 | 176.55 |
| Donor (i) | Occupancy(e) | Acceptor (j) | Occupancy(e) | E(2) a kcal/mol | E(j) – E(i) b (a.u) | F(i,j) c (a.u) |
|---|---|---|---|---|---|---|
| π(C1–C6) | 1.70639 | π*(C2–C3) | 0.36424 | 20.27 | 0.29 | 0.070 |
| π(C1–C6) | 1.70639 | π*(C4–C5) | 0.37847 | 17.86 | 0.30 | 0.067 |
| π(C2–C3) | 1.69340 | π*(C1–C6) | 0.33036 | 18.94 | 0.30 | 0.068 |
| π(C2–C3) | 1.69340 | π*(C4–C5) | 0.37847 | 19.07 | 0.30 | 0.069 |
| π(C4–C5) | 1.69274 | π*(C1–C6) | 0.33036 | 17.00 | 0.29 | 0.063 |
| π(C4–C5) | 1.69274 | π*(C2–C3) | 0.36424 | 17.47 | 0.28 | 0.063 |
| π(C4–C5) | 1.69274 | π*(C10–N18) | 0.18707 | 17.62 | 0.28 | 0.064 |
| π(C14–C15) | 1.85899 | π*(N16–C17) | 0.37926 | 14.88 | 0.28 | 0.061 |
| LP2(O7) | 1.85977 | π*(C1–C6) | 0.33036 | 25.80 | 0.36 | 0.090 |
| LP2(O9) | 1.85270 | π*(C2–C3) | 0.36424 | 27.07 | 0.35 | 0.093 |
| LP1(N13) | 1.55644 | π*(C14–C15) | 0.30606 | 30.67 | 0.29 | 0.087 |
| LP1(N13) | 1.55644 | π*(N16–C17) | 0.37926 | 46.28 | 0.28 | 0.103 |
| LP1(N18) | 1.95533 | σ*(C10–C11) | 0.03261 | 8.70 | 0.83 | 0.076 |
| LP1(O19) | 1.99160 | σ*(C11–H25) | 0.01285 | 0.67 | 1.11 | 0.024 |
| LP2(O19) | 1.90883 | π*(C10–N18) | 0.18707 | 15.97 | 0.35 | 0.068 |
| Wavenumber | Intensity | Assignment with PED % (≥10%) | |||
|---|---|---|---|---|---|
| Expt. | Calc. | IR a (km·mol–1) | Raman b (m2·sr−1) | ||
| νIR (cm–1) | νRaman (cm–1) | νScal (cm–1) | |||
| 3830 | 131.118 | 7.45 | ν (O19–H32) (100) | ||
| 3144 | 3145 | 3128 | 1.497 | 8.37 | νring II (C–H) (99) |
| 3121 | 3120 | 3118 | 2.054 | 7.851 | νring II (C–H) (99) |
| 3114 | 3118 | 3103 | 4.324 | 10.20 | νring II (C–H) (99) |
| - | 3073 | 3096 | 3.035 | 6.57 | νring II (C–H) (99) |
| 3018 | - | 3056 | 1.286 | 5.39 | νring I (C–H) (99) |
| 3007 | 3001 | 3026 | 2.149 | 10.90 | νring I (C–H) (99) |
| 2993 | - | 3017 | 3.215 | 11.70 | νring I (C–H) (99) |
| - | 2970 | 2977 | 9.230 | 2.62 | νas (CH2) Me III(93) |
| 2945 | 2946 | 2944 | 29.934 | 17.60 | νas (CH2) Me I (75) + νs (CH2) Me I (25) |
| - | - | 2934 | 23.671 | 11.10 | νs (CH2) Me III (98) |
| - | - | 2927 | 4.531 | 7.86 | νas (CH2) Me II (93) |
| - | 2908 | 2889 | 6.873 | 10.60 | νs (CH2) Me II (98) |
| - | 2845 | 2836 | 118.557 | 27.20 | νs (CH2) Me I (74) + νas (CH2) Me I (26) |
| - | 1629 | 1629 | 14.078 | 73.10 | ν (C10–N18) (71) |
| 1595 | 1606 | 1591 | 24.766 | 100.00 | νring I (CC) (53) + β (CH) Ring I (16) |
| - | - | 1581 | 3.985 | 20.60 | νring I (CC) (58) + β (CH) Ring I (14) |
| - | - | 1560 | 0.336 | 14.40 | Sci(HC) Me I (89) |
| 1503 | 1516 | 1504 | 44.673 | 2.85 | β (CH) Ring II (41) + ν (C17–N16) (28) +ν (NC) (12) |
| - | - | 1492 | 179.197 | 6.28 | β (CH) Ring I (50) + νring I (CC) (32) |
| 1493 | 1494 | 1490 | 17.736 | 6.84 | ν (C14–C15) (35) + β (CH) Ring II (29) +ν (NC) (11) |
| 1450 | - | 1457 | 14.740 | 4.35 | Sci (CH2) Me II (37) + τ (CN) (31) + Sci (CH2) Me III (16) |
| - | - | 1444 | 3.326 | 21.40 | ω (CH2) Me I (88) |
| 1439 | 1440 | 1442 | 25.303 | 19.50 | Sci (CH2) Me II + τ (CN) (25) + Sci (CH2) Me III (14) |
| 1428 | - | 1424 | 55.376 | 5.77 | νring I (CC) (44) + β (CH) Ring I (34) |
| 1401 | 1403 | 1390 | 17.021 | 13.50 | τ (CN) (47) + ω (CH2) Me III (30) |
| 1364 | - | 1369 | 2.376 | 5.68 | ν (NC) (28) + τ (CN) (17) + ω (CH2) Me III (12) + ρ (CH2) Me III (12) + twi (CH2) Me III (12) |
| 1350 | 1346 | 1341 | 2.477 | 15.10 | β (CH) Ring II (39) + ν (C17–N16) (31) + ν (NC) (15) |
| 1313 | 1314 | 1324 | 38.626 | 10.90 | β (CH) Ring I (40) + ν (CC) (37) + δa (Ring I) (10) |
| - | - | 1301 | 90.153 | 46.50 | νring I (CC) (32) + β (CH) Ring I (15) |
| 1280 | 1291 | 1293 | 9.747 | 28.50 | β (CH) Ring II (55) + ν (C17–N16) (16) |
| - | 1271 | 1276 | 18.733 | 10.70 | ω (CH2) Me II (62) |
| 1255 | 1254 | 1251 | 4.921 | 3.83 | Twi (CH2) Me II (34) + τ (CN) (24) + ρ (CH2) Me III (10) + Twi (CH2) Me III (10) |
| - | - | 1225 | 26.089 | 28.90 | ω (CH2) Me I (37) |
| 1225 | 1232 | 1224 | 199.125 | 31.10 | β (NOH) (23) + CO (13) + CC (12) |
| - | - | 1222 | 78.688 | 30.70 | ν (NC) (16) + CN (13) + β (CH) Ring II (13) + Twi (CH2) Me I (11) |
| - | - | 1208 | 103.083 | 19.30 | β (NOH) (23) + νring I (CC) (16) + ν (CO) (10) + β (CH) Ring I (10) |
| - | - | 1202 | 120.699 | 19.90 | νring I (CC) (30) + ν (C5–C10) (10) + β (NOH) (10) |
| 1173 | 1174 | 1151 | 4.028 | 2.22 | νring I (CC) (25) + β (CH) Ring I (25) |
| 1147 | 1141 | 1136 | 6.792 | 2.61 | ν (NC) (28) + Twi (CH2) Me I (14) + νring I (CC) (14) + β (CH) Ring I (12) |
| - | - | 1124 | 5.682 | 2.44 | ρ (CH2) Me I (56) + τ (CO) (10) + νring I (CC) (10) |
| 1108 | 1107 | 1111 | 12.714 | 7.30 | ν (NC) (32) + β (CH) Ring II (25) + ν (C14–C15) (11) |
| 1085 | 1083 | 1097 | 54.779 | 15.00 | δ (Ring I) (20) + ν (CO) (16) + ν (NC) (14) |
| - | 1050 | 1079 | 51.301 | 13.80 | β (CH) Ring II (43) + ν (NC) (32) + ν (C14–C15)(15) |
| 1035 | 1030 | 1032 | 12.857 | 19.20 | τ (CN) (25) + ρ (CH2) Me III (21) + Twi (CH2) Me III (21) |
| 1016 | - | 1019 | 121.778 | 4.74 | ν (OC) (36) + τ (CN) (11) |
| 957 | 956 | 1000 | 50.386 | 9.32 | ν (CC) (26) + νring I (CC) (14) |
| - | - | 972 | 6.193 | 14.00 | ν (CC)(66) |
| 936 | - | 966 | 16.460 | 14.90 | δ (Ring I) (34) + ν (NC) (27) + ν (CN) (13) |
| - | - | 935 | 3.579 | 3.29 | ω (CH) Ring I (86) |
| 924 | 922 | 921 | 100.257 | 9.39 | ν (N18–O19) (34) + τ (CN) (14) +ρ (CH2) Me III (10) + Twi (CH2) Me III (10) |
| 895 | 893 | 896 | 54.208 | 6.21 | ν (CO) (70) + δ (Ring I) (14) |
| 878 | - | 883 | 61.051 | 8.07 | ν (CO) (24) + ν (N18–O19) (16) + δ (Ring I) (13) |
| 854 | 850 | 851 | 24.434 | 2.27 | ω (CH) Ring I (77) |
| - | - | 841 | 2.112 | 2.68 | ω (CH) Ring II(87) |
| - | - | 818 | 7.104 | 4.86 | γ (Ring II) (89) |
| 808 | 814 | 809 | 21.360 | 7.71 | ω (CH) Ring I (84) |
| 799 | - | 798 | 13.387 | 28.60 | νring I (CC) (33) + ν (CO) (18) + ν (CO) (13) |
| 752 | - | 785 | 32.319 | 4.73 | ω (CH) Ring II (77) + ORO (11) |
| 743 | - | 756 | 2.956 | 6.19 | τ (CN) (40) + ρ (CH2) Me III (17) + twi (CH2) Me III (17) |
| - | - | 743 | 2.742 | 5.72 | τ (CN) (26) + ρ (CH2) Me III (11) + twi (CH2) Me III (11) |
| - | - | 733 | 9.478 | 12.10 | τ (CN) (37) + ρ (CH2) Me III (15) + twi (CH2) Me III (15) |
| 725 | 721 | 706 | 33.336 | 14.00 | ω (CH) Ring II (82) + τ (CN) (14) |
| 716 | - | 704 | 3.019 | 15.40 | γ (Ring I) (18) + δ (Ring I) (11) + γ (Ring I) (11) |
| - | - | 690 | 5.722 | 5.12 | puc (Ring I) (61) + ω (CC) (13) + τa (Ring I) (10) |
| - | 692 | 650 | 5.478 | 2.91 | ω (CC) (15) + τa (Ring I) (12) + τ (Ring I) (10) |
| 658 | 658 | 629 | 17.050 | 1.48 | τa (Ring II) (69) + τ (CN) (17) |
| 621 | 626 | 614 | 18.672 | 4.91 | δ (Ring II) (17) + ν (C12–N13) (15) + ν (CC) (11) |
| 578 | 573 | 592 | 11.347 | 7.42 | τa (Ring II) (62) + ω (CC) (11) |
| 556 | - | 582 | 14.299 | 9.94 | δ (Ring I) (30) |
| 492 | 490 | 530 | 5.947 | 2.11 | τ (CN) (20) + ω (CC) (14) + puc (14) |
| - | - | 447 | 5.385 | 7.04 | γ (Ring I) (20) + OC (19) + δ (Ring I) (15) |
| 433 | 449 | 440 | 19.054 | 7.96 | β (CNO) (21) + δ (Ring I) (15) + ν (CC) (10) |
| 425 | - | 424 | 86.609 | 9.89 | τ (NO) (72) |
| 417 | - | 405 | 13.261 | 4.70 | τa (Ring I) (36) + butt (35) + τ (Ring I) (14) |
| - | 387 | 361 | 1.486 | 7.82 | τ (CN) (26) + β (CN) (20) + τ (CN) (10) |
| - | - | 341 | 1.034 | 10.40 | τ (CN) (30) + Sci (13) + OC (12) |
| - | 330 | 333 | 0.753 | 9.09 | τ (CN) (67) + β (CN) (11) |
| - | 303 | 303 | 4.676 | 15.80 | τ (CN) (38) + τ (CN) (15) + butt (11) |
| - | 277 | 260 | 1.201 | 20.60 | τ (CN) (69) |
| - | 230 | 222 | 0.661 | 34.50 | τ (CN) (51) + ρ (OC) (10) |
| - | - | 215 | 0.930 | 29.90 | τa (Ring II) + (24) + τ (Ring I) (24) |
| - | - | 180 | 0.384 | 13.60 | τ (CN) (73) + β (CC) (10) |
| - | - | 138 | 0.183 | 22.60 | τ (CN) (37) + Sci (20) |
| - | - | 126 | 7.961 | 18.10 | τ (Ring I) (40) + τ (CN)(18) + τ (CN) (18) + τ (CN) (14) |
| - | - | 91 | 5.187 | 119.00 | τ (CN) (27) + τ (Ring I) (17) + ω (CC) (12) + τ (CN)(10) |
| - | - | 72 | 1.075 | 156.00 | τ (CN) (99) |
| - | - | 64 | 0.692 | 162.00 | τ (CN) (78) |
| - | - | 29 | 1.601 | 104.00 | τ (CN) (98) |
| - | - | 20 | 1.176 | 312.00 | τ (CN) (99) |
| - | - | 16 | 0.169 | 328.00 | τ (CN) (86) |
| Carbon Atoms (13C) | Hydrogen Atoms (1H) | ||||
|---|---|---|---|---|---|
| Atoms | Value (ppm) | Atoms | Value (ppm) | ||
| Calc. | Exp. | Calc. | Exp. | ||
| C1 | 127.25 | 148.0 | H20 | 5.81 | 6.84 |
| C2 | 127.81 | 148.3 | H21 | 7.01 | 7.06 |
| C3 | 89.62 | 108.5 | H22 | 5.89 | 7.13 |
| C4 | 107.84 | 120.5 | H23 | 5.40 | 6.04 |
| C5 | 107.85 | 130.3 | H24 | 5.20 | 6.04 |
| C6 | 89.80 | 106.1 | H25 | 1.92 | 3.14 |
| C8 | 87.78 | 101.7 | H26 | 2.23 | 3.14 |
| C10 | 132.24 | 153.7 | H27 | 3.40 | 4.17 |
| C11 | 29.66 | 28.2 | H28 | 3.64 | 4.17 |
| C12 | 38.54 | 43.3 | H29 | 6.05 | 6.89 |
| C14 | 100.78 | 119.9 | H30 | 5.94 | 7.19 |
| C15 | 109.30 | 128.3 | H31 | 6.38 | 7.66 |
| C17 | 115.68 | 137.5 | |||
| Compound No. | MIC (μmol/L) | |||
|---|---|---|---|---|
| C. albicans | C. tropicalis | C.parapsilosis | Aspergillus niger | |
| 4 | 987.43 | 987.43 | 987.43 | 987.43 |
| Ketoconazole | 7.53 | 15.05 | 15.05 | 15.05 |
| Chemical Formula | 2(C13H13N3O3)·C3H8O |
|---|---|
| Molecular weight | 578.62 |
| Crystal system, space group | Monoclinic, P21 |
| Temperature (K) | 296 |
| a, b, c (Å) | 9.0963(3), 14.7244(6), 10.7035(4) |
| β(°) | 94.298(3) |
| V (Å3) | 1429.57(9) |
| Z | 2 |
| Radiation type | Cu Kα |
| µ (mm–1) | 0.81 |
| Crystal size (mm) | 0.40 × 0.23 × 0.11 |
| Data collection | |
| Diffractometer | Bruker APEX-II CCD diffractometer |
| Absorption correction | Multi-scan, SADABS Bruker 2014 |
| Tmin, Tmax | 0.740, 0.916 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 6654, 3851, 2788 |
| Rint | 0.039 |
| Refinement | |
| R[F2> 2σ(F2)] a, wR(F2) b, S | 0.059, 0.144, 1.03 |
| No. of reflections | 3851 |
| No. of parameters | 393 |
| No. of restraints | 1 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e·Å–3) | 0.26, −0.23 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wabli, R.I.; Al-Ghamdi, A.R.; Ghabbour, H.A.; Al-Agamy, M.H.; Monicka, J.C.; Joe, I.H.; Attia, M.I. Synthesis, X-ray Single Crystal Structure, Molecular Docking and DFT Computations on N-[(1E)-1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A New Potential Antifungal Agent Precursor. Molecules 2017, 22, 373. https://doi.org/10.3390/molecules22030373
Al-Wabli RI, Al-Ghamdi AR, Ghabbour HA, Al-Agamy MH, Monicka JC, Joe IH, Attia MI. Synthesis, X-ray Single Crystal Structure, Molecular Docking and DFT Computations on N-[(1E)-1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A New Potential Antifungal Agent Precursor. Molecules. 2017; 22(3):373. https://doi.org/10.3390/molecules22030373
Chicago/Turabian StyleAl-Wabli, Reem I., Alwah R. Al-Ghamdi, Hazem A. Ghabbour, Mohamed H. Al-Agamy, James Clemy Monicka, Issac Hubert Joe, and Mohamed I. Attia. 2017. "Synthesis, X-ray Single Crystal Structure, Molecular Docking and DFT Computations on N-[(1E)-1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A New Potential Antifungal Agent Precursor" Molecules 22, no. 3: 373. https://doi.org/10.3390/molecules22030373
APA StyleAl-Wabli, R. I., Al-Ghamdi, A. R., Ghabbour, H. A., Al-Agamy, M. H., Monicka, J. C., Joe, I. H., & Attia, M. I. (2017). Synthesis, X-ray Single Crystal Structure, Molecular Docking and DFT Computations on N-[(1E)-1-(2H-1,3-Benzodioxol-5-yl)-3-(1H-imidazol-1-yl)propylidene]-hydroxylamine: A New Potential Antifungal Agent Precursor. Molecules, 22(3), 373. https://doi.org/10.3390/molecules22030373