
High Hydrostatic Pressure (HHP)-Induced Structural Modification of Patatin and Its Antioxidant Activities


Abstract
:1. Introduction
2. Results and Discussion
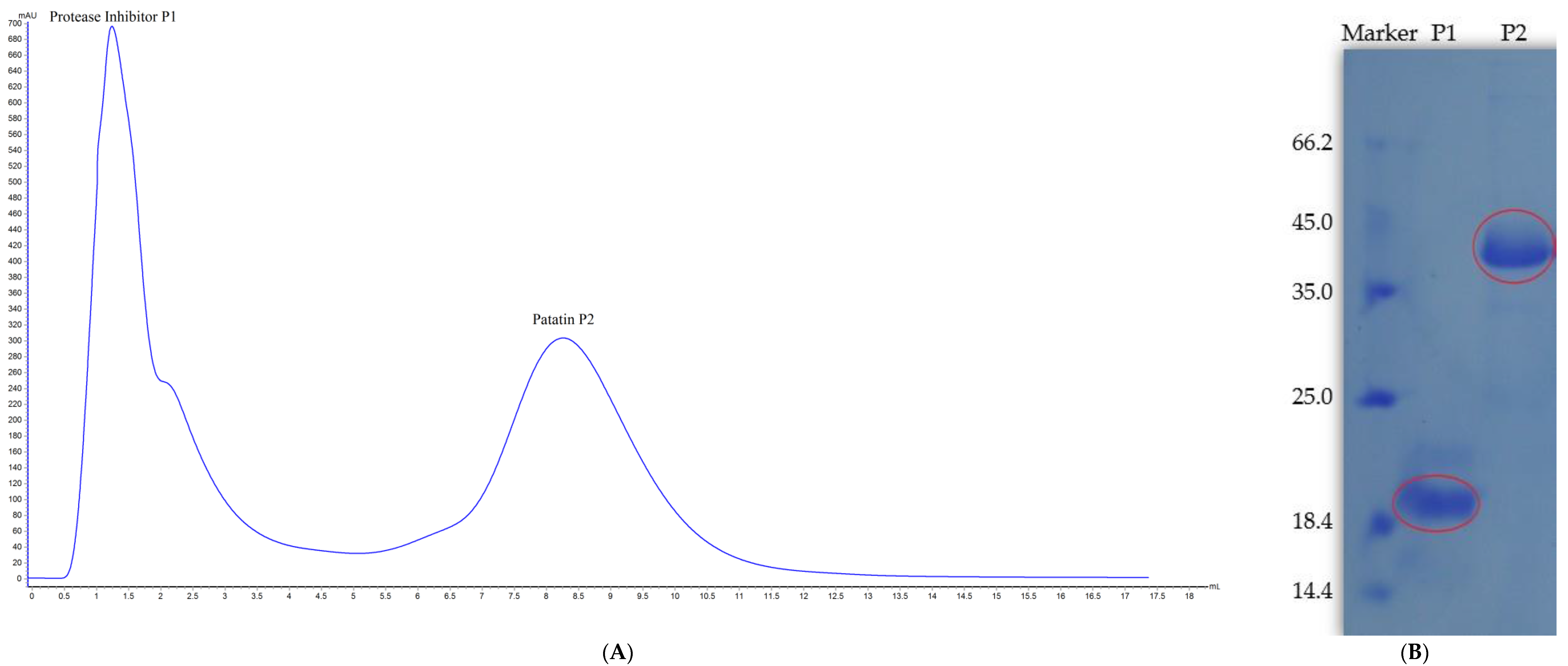
2.1. Patatin Purification
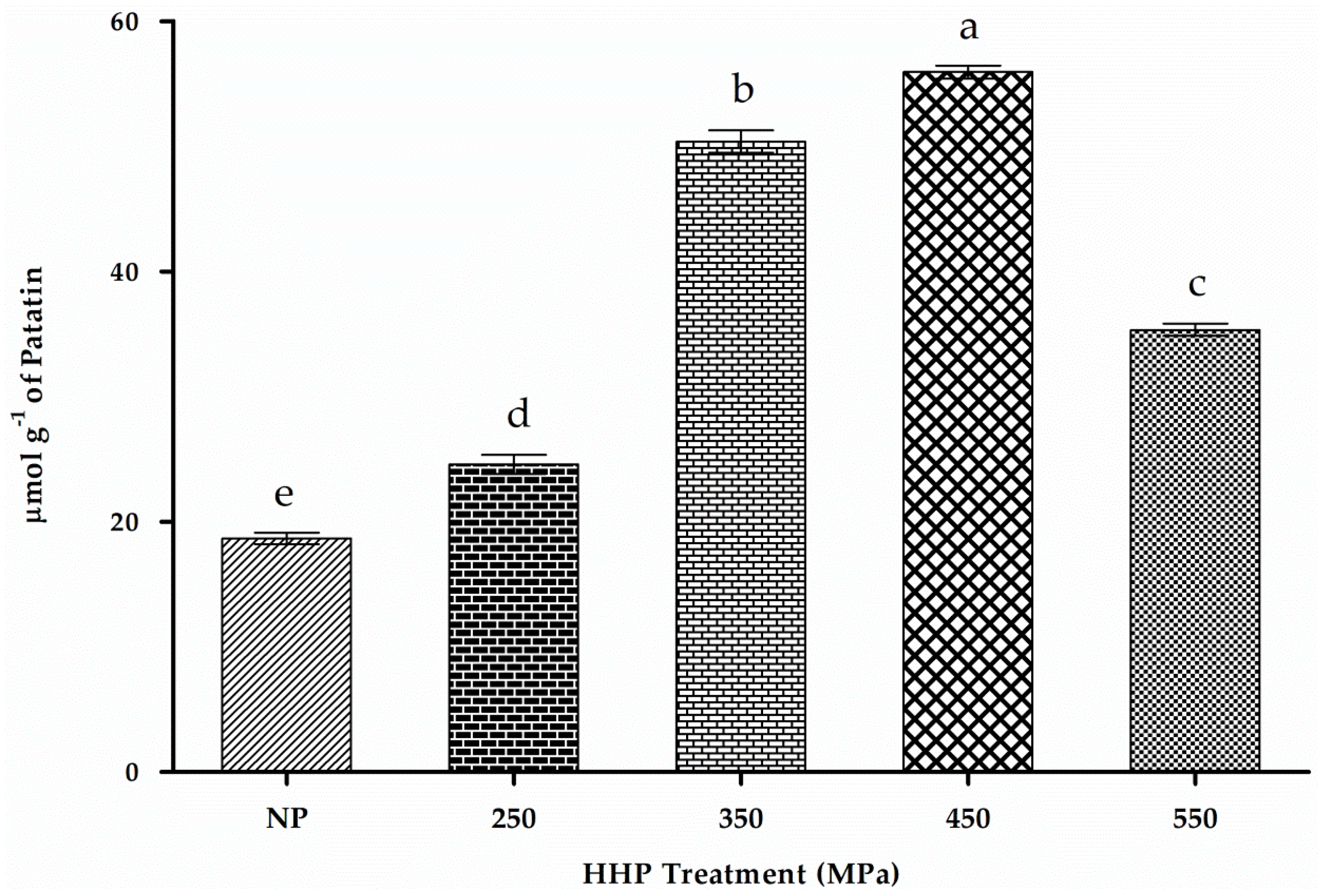
2.2. Patatin Yield and Purity
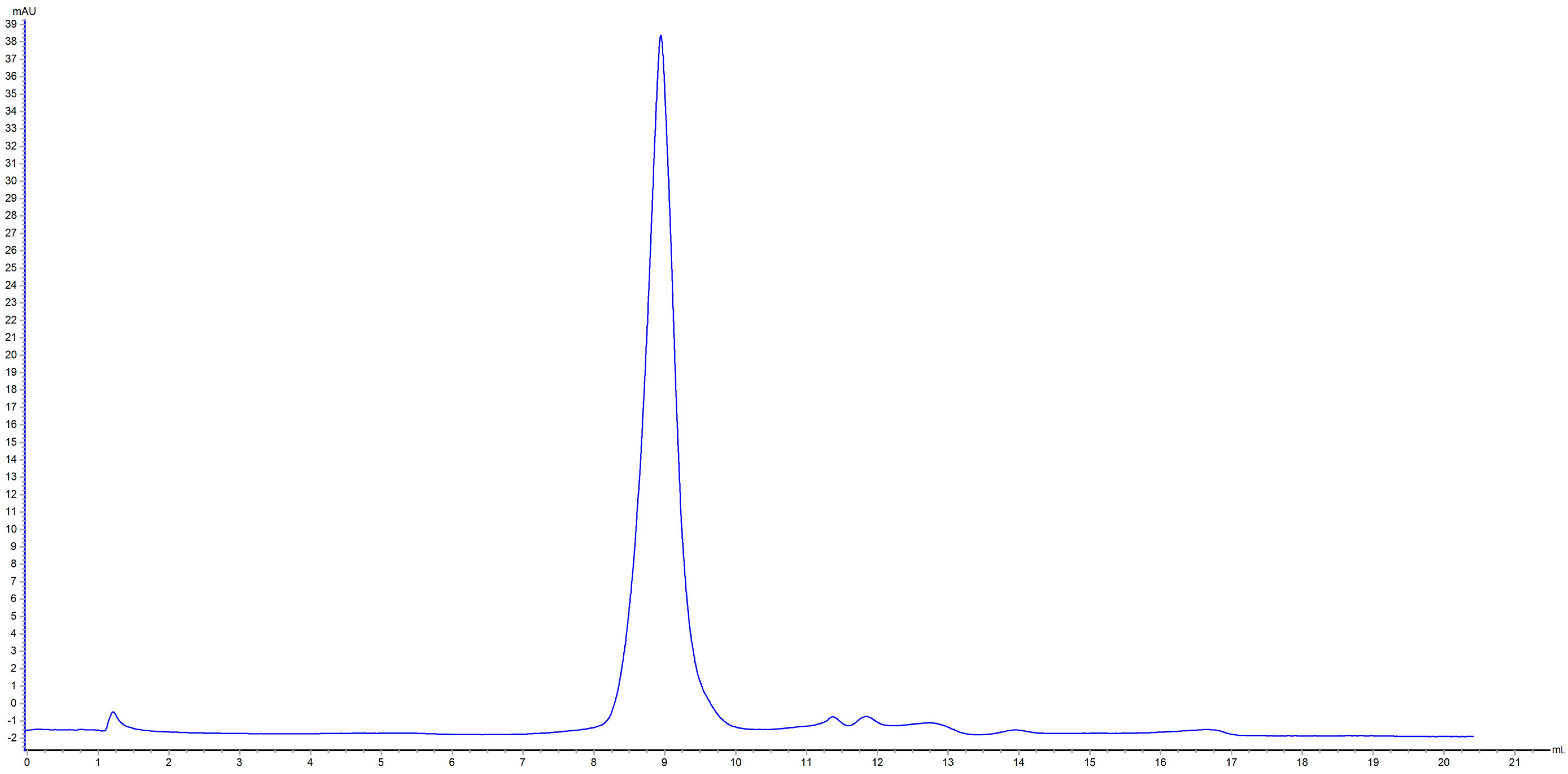
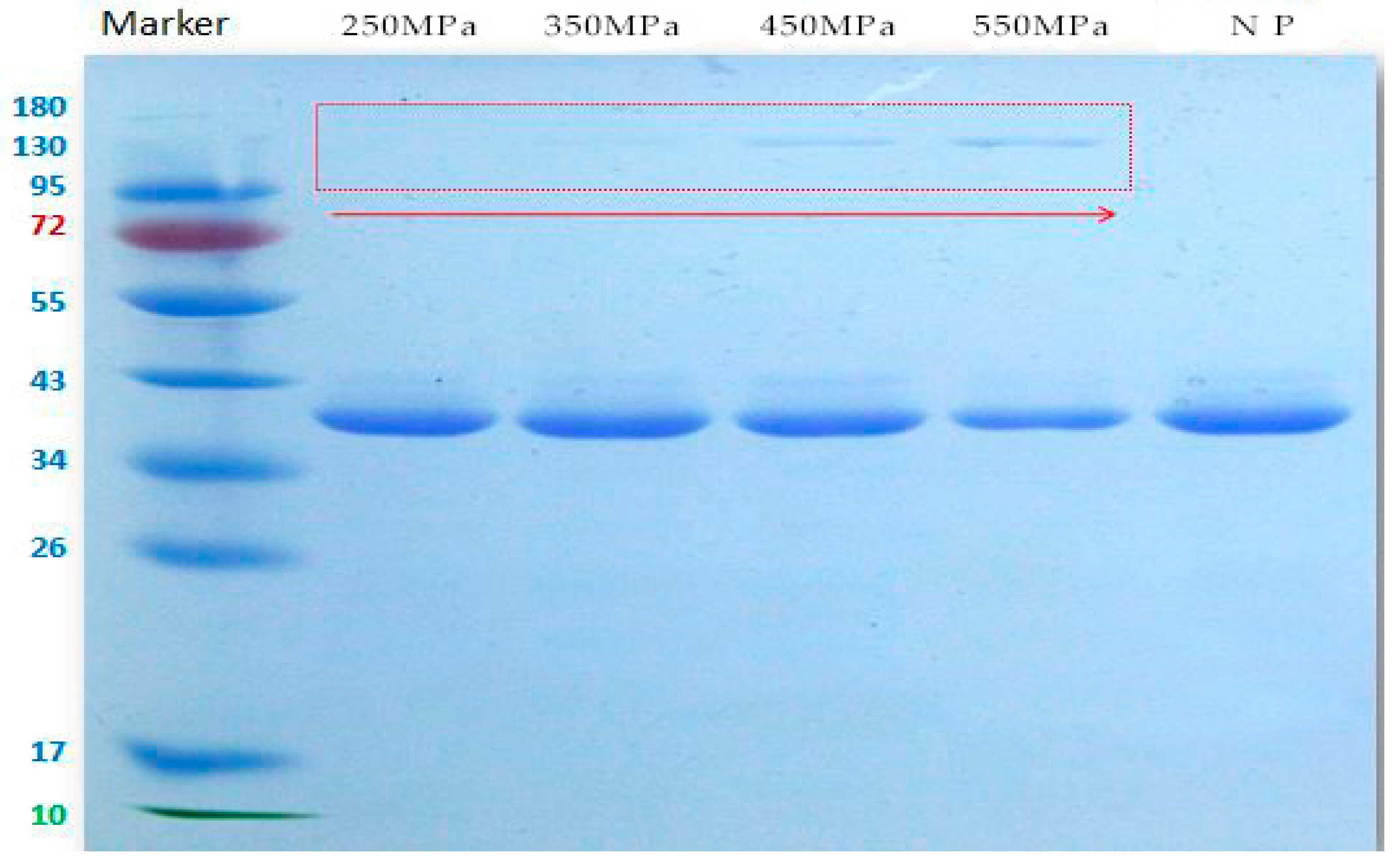
2.3. Effects of HHP on MW Distribution of Patatin
2.4. Neutral Sugar Composition
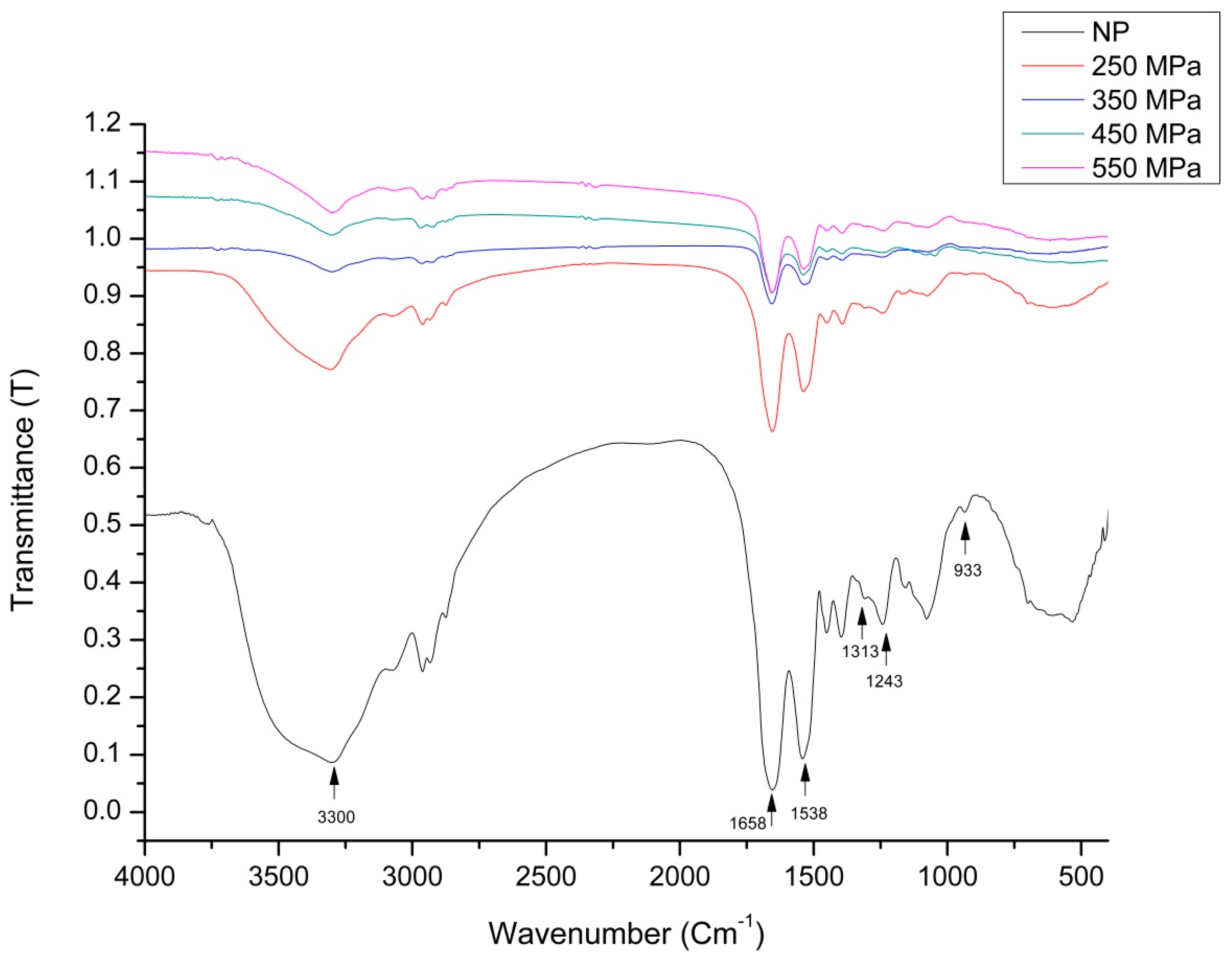
2.5. FTIR Analysis
2.6. DSC
2.7. CD
2.8. Ho
2.9. Free-SH
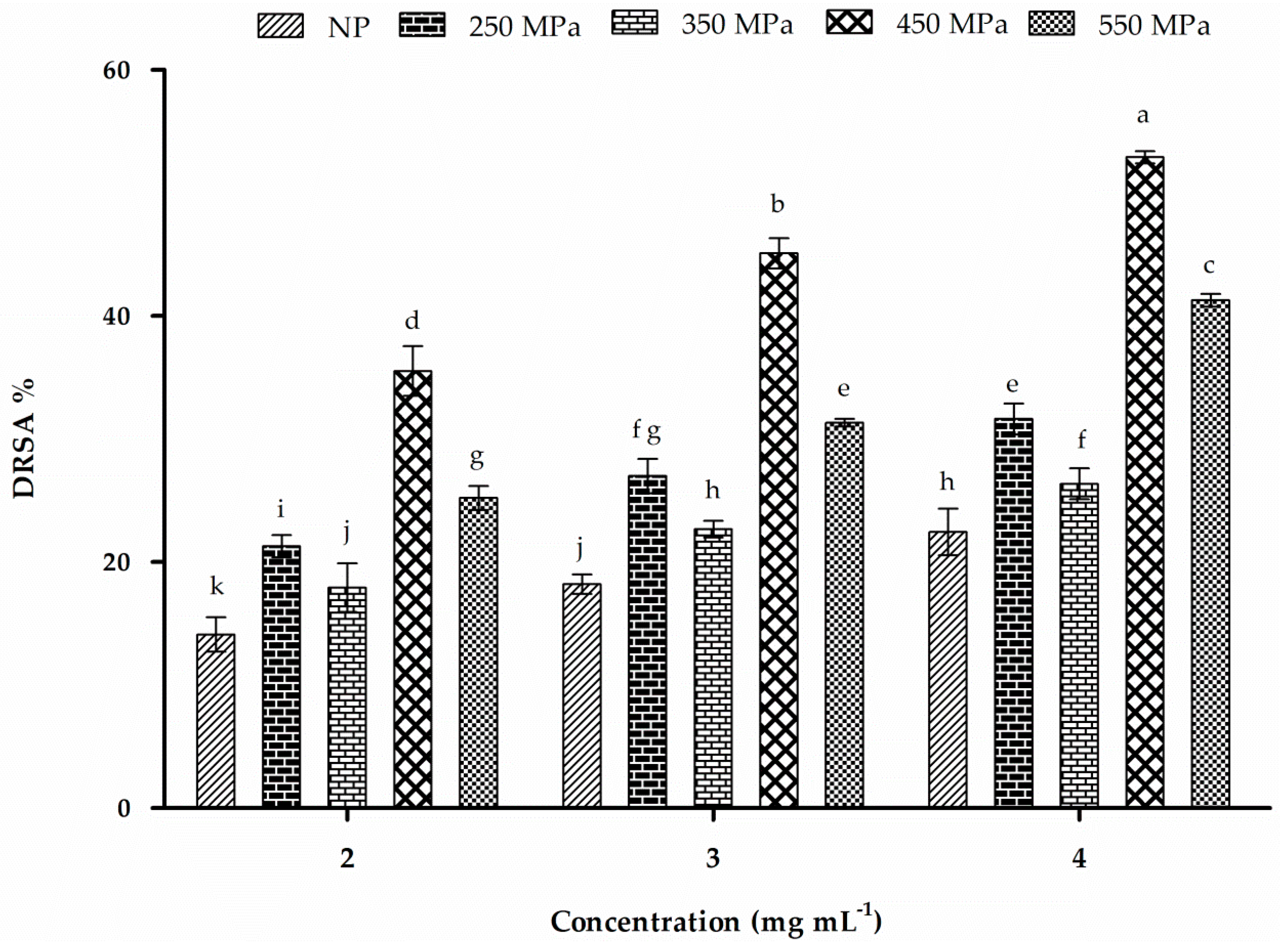
2.10. DPPH-Radical-Scavenging Activity
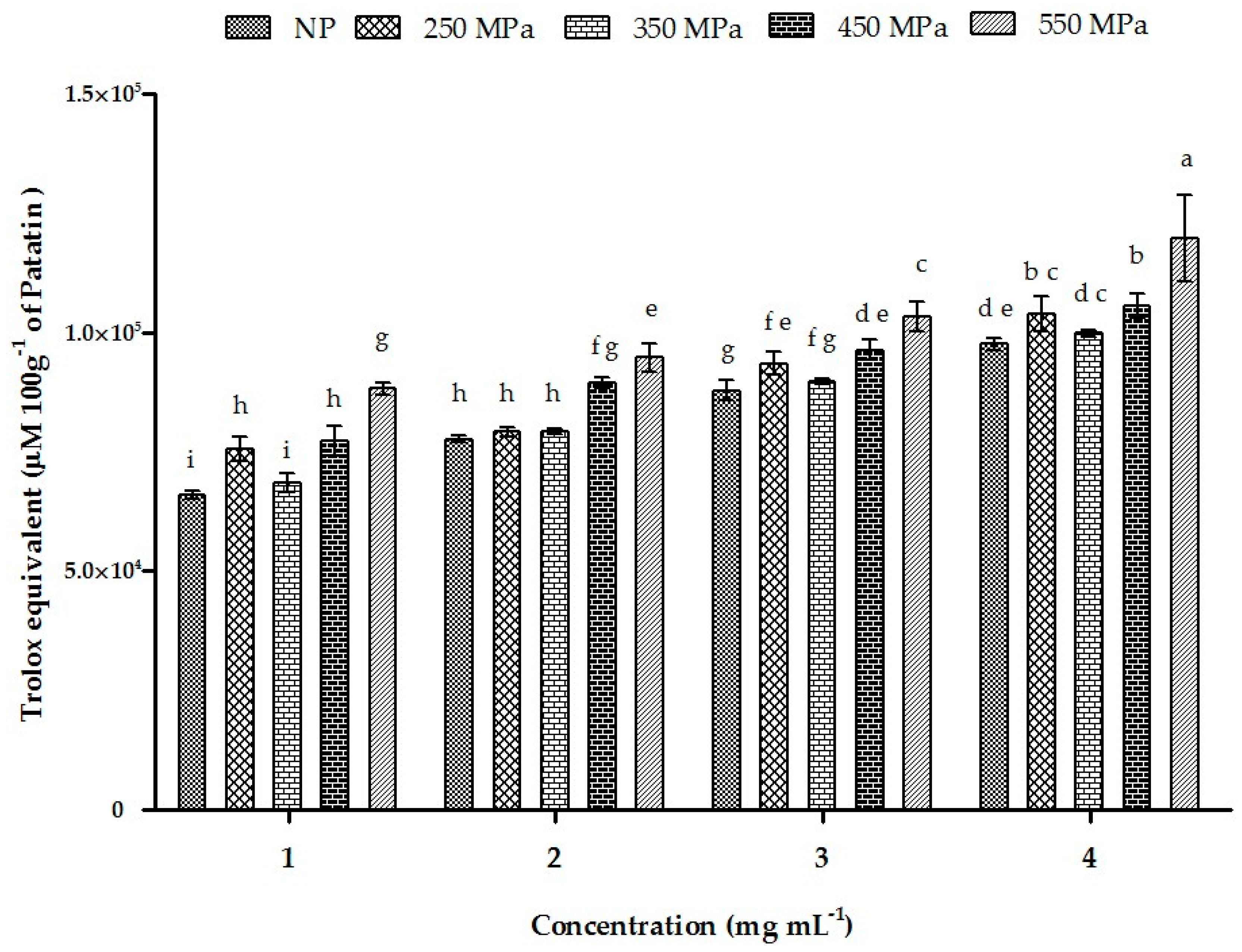
2.11. ORAC
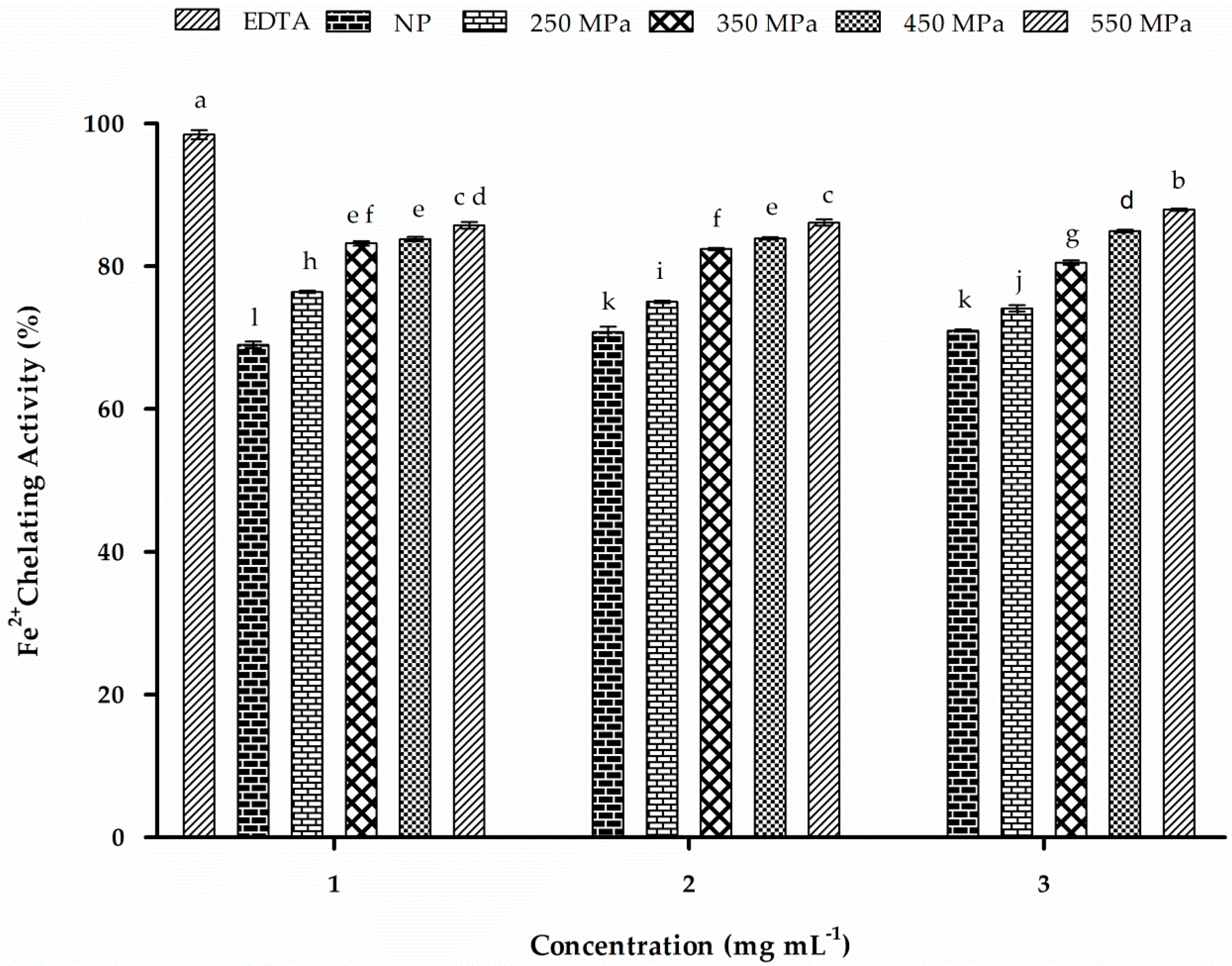
2.12. Iron-Chelating Activity
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Patatin Isolation and Purification
3.3. High-Pressure Treatment of Patatin Samples
3.4. Patatin MW Determination
3.5. Neutral Sugar Composition of Patatin
3.6. Fourier Transform Infrared Spectroscopy (FTIR) Spectrum of Patatin
3.7. Differential Scanning Calorimetry (DSC)
3.8. Circular Dichroism (CD)
3.9. Surface Hydrophobicity (Ho)
3.10. Determination of Free Sulfhydryl Groups
3.11. DPPH-Radical-Scavenging Activity Assay
3.12. Oxygen-Radical Absorbance Capacity (ORAC) Assay
3.13. Ferrous Ion-Chelating Activity
3.14. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- FAOSTAT. Food and Nutrition; FAO: Rome, Italy, 2014. [Google Scholar]
- Du-qin, Z.; Tai-hua, M.U.; Hong-nan, S.U.N. Domestic and Abroad Research Progress of Potato Tuber-Specific Storage Protein Patatin. Sci. Agric. Sin. 2016, 49, 1746–1756. [Google Scholar]
- Shewry, P.R. Tuber storage proteins. Ann. Bot. 2003, 91, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Pots, A.M.; Gruppen, H.; Van Diepenbeek, R.; Van Der Lee, J.J.; Van Boekel, M.A.J.S.; Wijngaards, G.; Voragen, A.G.J. The effect of storage of whole potatoes of three cultivars on the patatin and protease inhibitor content; a study using capillary electrophoresis and MALDI-TOF mass spectrometry. J. Sci. Food Agric. 1999, 79, 1557–1564. [Google Scholar] [CrossRef]
- Bárta, J.; Bártová, V. Patatin, the major protein of potato (Solanum tuberosum L.) tubers, and its occurrence as genotype effect: Processing versus table potatoes. Czech J. Food Sci. 2008, 26, 347–359. [Google Scholar]
- Racusen, D.; Foote, M. A Major Soluble Glycoprotein of Potato Tubers. J. Food Biochem. 1980, 4, 43–52. [Google Scholar] [CrossRef]
- Pouvreau, L.; Gruppen, H.; Piersma, S.R.; van den Broek, L.A.; van Koningsveld, G.A.; Voragen, A.G. Relative abundance and inhibitory distribution of protease inhibitors in potato juice from cv. Elkana. J. Agric. Food Chem. 2001, 49, 2864–2874. [Google Scholar] [CrossRef] [PubMed]
- Man, A.L.; Purcell, P.C.; Hannappel, U.; Halford, N.G. Potato SNF1-related protein kinase: Molecular cloning, expression analysis and peptide kinase activity measurements. Plant Mol. Biol. 1997, 34, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Racusen, D.; Weller, D.L. Molecular weight of patatin, A major potato tuber protein. J. Food Biochem. 1984, 8, 103–107. [Google Scholar] [CrossRef]
- Pots, A. Physico-chemical Properties and Thermal Aggregation of Patatin, the Major Potato Tuber Protein; Landbouwuniversiteit: Wageningen, The Netherlands, 1999. [Google Scholar]
- Sun, Y.; Jiang, L.; Wei, D. Partial characterization, in vitro antioxidant and antiproliferative activities of patatin purified from potato fruit juice. Food Funct. 2013, 4, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Giuseppin, M.L.F.; Van Der Sluis, C.; Laus, M.C. Native Potato Protein Isolate. U.S. Patent US2013/0281669 A1, 18 February 2010. [Google Scholar]
- Waglay, A.; Karboune, S.; Alli, I. Potato protein isolates: Recovery and characterization of their properties. Food Chem. 2014, 142, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-W.; Han, C.-H.; Lee, M.-H.; Hsu, F.-L.; Hou, W.-C. Patatin, the tuber storage protein of potato (Solanum tuberosum L.), exhibits antioxidant activity in vitro. J. Agric. Food Chem. 2003, 51, 4389–4393. [Google Scholar] [CrossRef] [PubMed]
- Barrio, D.A.; Anon, M.C. Potential antitumor properties of a protein isolate obtained from the seeds of Amaranthus mantegazzianus. Eur. J. Nutr. 2010, 49, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Al-Saikhan, M.S.; Howard, L.R.; Miller, J.C., Jr. Antioxidant Activity and Total Phenolics in Different Genotypes of Potato (Solanum tuberosum, L.). J. Food Sci. 1995, 60, 341–343. [Google Scholar] [CrossRef]
- Galliard, T. The enzymic deacylation of phospholipids and galactolipids in plants. Purification and properties of a lipolytic acyl-hydrolase from potato tubers. Biochem. J. 1971, 121, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2008, 50, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, N.K.; Raghavarao, K.S.; Balasubramaniam, V.M.; Niranjan, K.; Knorr, D. Opportunities and Challenges in High Pressure Processing of Foods. Crit. Rev. Food Sci. Nutr. 2007, 47, 69–112. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, A. Examination of Egg White Proteins and Effects of High Pressure on Select Physical and Functional Properties. Master’s Thesis, University of Nebraska Lincoln, Lincoln, NE, USA, 2010. [Google Scholar]
- Sun, X.D.; Holley, R.A. High hydrostatic pressure effects on the texture of meat and meat products. J. Food Sci. 2010, 75, R17–R23. [Google Scholar] [CrossRef] [PubMed]
- Neetoo, H.; Chen, H. Application of High Hydrostatic Pressure Technology for Processing and Preservation of Foods. In Progress in Food Preservation; Bhat, R., Alias, A.K., Paliyath, G., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2012; pp. 247–276. [Google Scholar]
- Balci, A.T.; Wilbey, R.A. High pressure processing of milk-the first 100 years in the development of a new technology. Int. J. Dairy Technol. 1999, 52, 149–155. [Google Scholar] [CrossRef]
- Phillips, G.O.; Williams, P.A. (Eds.) Handbook of Food Proteins; Woodhead Publishing Limited: Cornwall, UK, 2011.
- AOAC. Official Methods of Analysis of AOAC International, 15th ed.; Helrich, K., Ed.; AOAC International: Gaithersburg, MD, USA, 1990. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurment With the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Jørgensen, M.; Bauw, G.; Welinder, K.G. Molecular properties and activities of tuber proteins from starch potato cv. Kuras. J. Agric. Food Chem. 2006, 54, 9389–9397. [Google Scholar] [CrossRef] [PubMed]
- Bauw, G.; Nielsen, H.V.; Emmersen, J.; Nielsen, K.L.; Jørgensen, M.; Welinder, K.G. Patatins, Kunitz protease inhibitors and other major proteins in tuber of potato cv. Kuras. FEBS J. 2006, 273, 3569–3584. [Google Scholar] [CrossRef] [PubMed]
- He, R.; He, H.Y.; Chao, D.; Ju, X.; Aluko, R. Effects of High Pressure and Heat Treatments on Physicochemical and Gelation Properties of Rapeseed Protein Isolate. Food Bioprocess Technol. 2014, 7, 1344–1353. [Google Scholar] [CrossRef]
- Tang, C.-H.; Ma, C.-Y. Effect of high pressure treatment on aggregation and structural properties of soy protein isolate. LWT Food Sci. Technol. 2009, 42, 606–611. [Google Scholar] [CrossRef]
- Condés, M.C.; Speroni, F.; Mauri, A.; Añón, M.C. Physicochemical and structural properties of amaranth protein isolates treated with high pressure. Innov. Food Sci. Emerg. Technol. 2012, 14, 11–17. [Google Scholar] [CrossRef]
- Angsupanich, K.; Edde, M.; Ledward, D.A. Effects of high pressure on the myofibrillar proteins of cod and Turkey muscle. J. Agric. Food Chem. 1999, 47, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Liedl, B.E.; Kosier, T.; Desborough, S.L. HPLC Isolation and Nutritional Value of a Major Tuber Protein. Am. Potato J. 1987, 64, 545–557. [Google Scholar] [CrossRef]
- Lehesranta, S.J.; Davies, H.V.; Shepherd, L.V.T.; Nunan, N.; McNicol, J.W.; Auriola, S.; Koistinen, K.M.; Suomalainen, S.; Kokko, H.I.; Karenlampi, S.O. Comparison of tuber proteomes of potato varieties, landraces, and genetically modified lines. Plant Physiol. 2005, 138, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Pequeux, A.J.R.; Gilles, R. High Pressure Effects on Selected Biological Systems; Péqueux, A.J.R., Gilles, R., Eds.; Springer: Berlin/Heidelberg, Germany, 1985; Volume 53. [Google Scholar]
- Li, H.; Zhu, K.; Zhou, H.; Peng, W. Effects of high hydrostatic pressure treatment on allergenicity and structural properties of soybean protein isolate for infant formula. Food Chem. 2012, 132, 808–814. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J.; Bryce, D.L. Spectrometric Identification of Organic Compounds, 8th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 2015. [Google Scholar]
- Wang, X.S.; Tang, C.H.; Li, B.S.; Yang, X.Q.; Li, L.; Ma, C.Y. Effects of high-pressure treatment on some physicochemical and functional properties of soy protein isolates. Food Hydrocoll. 2008, 22, 560–567. [Google Scholar] [CrossRef]
- Chapleau, N.; Mangavel, C.; Compoint, J.P.; De Lamballerie-Anton, M. Effect of high-pressure processing on myofibrillar protein structure. J. Sci. Food Agric. 2004, 84, 66–74. [Google Scholar] [CrossRef]
- Larrea-Wachtendorff, D.; Tabilo-Munizaga, G.; Moreno-Osorio, L.; Villalobos-Carvajal, R.; Pérez-Won, M. Protein Changes Caused by High Hydrostatic Pressure (HHP): A Study Using Differential Scanning Calorimetry (DSC) and Fourier Transform Infrared (FTIR) Spectroscopy. Food Eng. Rev. 2015, 7, 222–230. [Google Scholar] [CrossRef]
- Katsura, H.; Zikihara, K.; Okajima, K.; Yoshihara, S.; Tokutomi, S. Oligomeric structure of LOV domains in Arabidopsis phototropin. FEBS Lett. 2009, 583, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Tabilo-Munizaga, G.; Gordon, T.A.; Villalobos-Carvajal, R.; Moreno-Osorio, L.; Salazar, F.N.; Pérez-Won, M.; Acuña, S. Effects of high hydrostatic pressure (HHP) on the protein structure and thermal stability of Sauvignon blanc wine. Food Chem. 2014, 155, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y.Y.; Li, M.-J.J.; Liang, R.-C.C.; Lee, S.-M.M. Non-destructive analysis of the conformational changes in human lens lipid and protein structures of the immature cataracts associated with glaucoma. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1998, 54, 1509–1517. [Google Scholar] [CrossRef]
- Kato, A.; Nakai, S. Hydrophobicity determined by a fluorescence probe method and its correlation with surface properties of proteins. Biochim. Biophys. Acta Protein Struct. 1980, 624, 13–20. [Google Scholar] [CrossRef]
- Alizadeh-Pasdar, N.; Li-Chan, E.C. Comparison of protein surface hydrophobicity measured at various pH values using three different fluorescent probes. J. Agric. Food Chem. 2000, 48, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Molina, E.; Papadopoulou, A.; Ledward, D.A. Emulsifying properties of high pressure treated soy protein isolate and 7S and 11S globulins. Food Hydrocoll. 2001, 15, 263–269. [Google Scholar] [CrossRef]
- Puppo, M.C.; Speroni, F.; Chapleau, N.; De Lamballerie, M.; Añón, M.C.; Anton, M. Effect of high-pressure treatment on emulsifying properties of soybean proteins. Food Hydrocoll. 2005, 19, 289–296. [Google Scholar] [CrossRef]
- Arpa-Parra, M.; Bamdad, F.; Tian, Z.; Zeng, H.; Temelli, F.; Chen, L. Impact of pH on molecular structure and surface properties of lentil legumin-like protein and its application as foam stabilizer. Colloids Surf. B Biointerfaces 2015, 132, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Kong, X.; Chen, Y.; Zhang, C.; Yang, Y.; Hua, Y. Effects of heat treatment on the emulsifying properties of pea proteins. Food Hydrocoll. 2016, 52, 301–310. [Google Scholar] [CrossRef]
- Alvarez, P.A.; Ramaswamy, H.S.; Ismail, A.A. High pressure gelation of soy proteins: Effect of concentration, pH and additives. J. Food Eng. 2008, 88, 331–340. [Google Scholar] [CrossRef]
- Mignery, G.A.; Pikaard, C.S.; Hannapel, D.J.; Park, W.D. Isolation and sequence analysis of cDNAs for the major potato tuber protein, patatin. Nucleic Acids Res. 1984, 12, 7987–8000. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, K.S. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry. J. Am. Chem. Soc. 1960, 82, 4121. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Tang, X.; Chen, Y.; You, Y. Chemical forces and water holding capacity study of heat-induced myofibrillar protein gel as affected by high pressure. Food Chem. 2015, 188, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Panick, G.; Malessa, R.; Winter, R. Differences between the pressure- and temperature-induced denaturation and aggregation of β-lactoglobulin A, B, and AB monitored by FT-IR spectroscopy and small-angle X-ray scattering. Biochemistry 1999, 38, 6512–6519. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Lin, L.; Liang, Y.; Benjakul, S.; Shi, X.; Liu, X. Physicochemical properties of natural actomyosin from threadfin bream (Nemipterus spp.) induced by high hydrostatic pressure. Food Chem. 2014, 156, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.M.; Sasaki, S.; McClements, D.J.; Decker, E.A. Mechanisms of the antioxidant activity of a high molecular weight fraction of whey. J. Agric. Food Chem. 2000, 48, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.-C.C.; Han, C.-H.H.; Chen, H.-J.J.; Wen, C.-L.L.; Lin, Y.-H.H. Storage proteins of two cultivars of sweet potato (Ipomoea batatas L.) and their protease hydrolysates exhibited antioxidant activity in vitro. Plant Sci. 2005, 168, 449–456. [Google Scholar] [CrossRef]
- Mozhaev, V.V.; Heremans, K.; Frank, J.; Masson, P.; Balny, C. High pressure effects on protein structure and function. Proteins 1996, 24, 81–91. [Google Scholar] [CrossRef]
- Haytowitz, D.; Bhagwat, S. USDA Database for the Oxygen Radical Absorbance Capacity (ORAC) of Selected Foods, Release 2; USDA: Beltsville, MD, USA, 2010. [Google Scholar]
- Zhu, K.; Zhou, H.; Qian, H. Antioxidant and free radical-scavenging activities of wheat germ protein hydrolysates (WGPH) prepared with alcalase. Process Biochem. 2006, 41, 1296–1302. [Google Scholar] [CrossRef]
- Tang, C.H.; Wang, X.S.; Yang, X.Q. Enzymatic hydrolysis of hemp (Cannabis sativa L.) protein isolate by various proteases and antioxidant properties of the resulting hydrolysates. Food Chem. 2009, 114, 1484–1490. [Google Scholar] [CrossRef]
- Peng, X.; Kong, B.; Xia, X.; Liu, Q. Reducing and radical-scavenging activities of whey protein hydrolysates prepared with Alcalase. Int. Dairy J. 2010, 20, 360–365. [Google Scholar] [CrossRef]
- Seppälä, U.; Alenius, H.; Turjanmaa, K.; Reunala, T.; Palosuo, T.; Kalkkinen, N. Identification of patatin as a novel allergen for children with positive skin prick test responses to raw potato. J. Allergy Clin. Immunol. 1999, 103, 165–171. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during Assembly of Head of Bacteriophage-T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Ogutu, F.O.; Mu, T.-H. Ultrasonic degradation of sweet potato pectin and its antioxidant activity. Ultrason. Sonochem. 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Tai-hua, M.; Han, J.-J. Composition and Physicochemical Properties of Dietary Fiber Extracted from Residues of 10 Varieties of Sweet Potato by a Sieving Method. J. Agric. Food Chem. 2010, 58, 7305–7310. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Mu, T.-H.; Zhang, M.; Chen, J. Effects of high hydrostatic pressure on the physicochemical and emulsifying properties of sweet potato protein. Int. J. Food Sci. Technol. 2013, 48, 1260–1268. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef]
- Paraman, I.; Hettiarachchy, N.S.; Schaefer, C. Preparation of rice endosperm protein isolate by alkali extraction. Cereal Chem. 2008, 85, 76–81. [Google Scholar] [CrossRef]
- Beveridge, T.; Toma, S.J.; Nakai, S. Determination of Sh- and Ss-Groups in Some Food Proteins Using Ellman’S Reagent. J. Food Sci. 1974, 39, 49–51. [Google Scholar] [CrossRef]
- Zhang, M.; Mu, T.; Wang, Y.; Sun, M. Evaluation of free radical-scavenging activities of sweet potato protein and its hydrolysates as affected by single and combination of enzyme systems. Int. J. Food Sci. Technol. 2012, 47, 696–702. [Google Scholar] [CrossRef]
- Prior, R.L.; Hoang, H.; Gu, L.; Wu, X.; Bacchiocca, M.; Howard, L.; Hampsch-Woodill, M.; Huang, D.; Ou, B.; Jacob, R. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORACFL)) of plasma and other biological and food samples. J. Agric. Food Chem. 2003, 51, 3273–3279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mu, T.-H.; Sun, M. Purification and identification of antioxidant peptides from sweet potato protein hydrolysates by Alcalase. J. Funct. Foods 2014, 7, 191–200. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Rhamnose | Galactose | Glucose | Xylose | Mannose |
|---|---|---|---|---|---|
| NP | 0.97 ± 0.01 c | 14.15 ± 0.64 d | 14.30 ± 0.25 a | 0.08 ± 0.00 a | 0.445 ± 0.02 b |
| 250 MPa | 1.69 ± 0.05 a | 18.04 ± 0.57 a | 10.93 ± 0.24 d | 0.08 ± 0.00 a | 0.563 ± 0.02 a |
| 350 MPa | 0.82 ± 0.03 d | 12.16 ± 0.17 e | 10.70 ± 0.08 e | 0.07 ± 0.00 b,c | 0.422 ± 0.01 b |
| 450 MPa | 0.97 ± 0.02 c | 14.27 ± 0.61 c | 12.30 ± 0.25 b | 0.07 ± 0.00 a,b | 0.467 ± 0.00 b |
| 550 MPa | 1.40 ± 0.03 b | 17.03 ± 1.13 b | 11.24 ± 0.37 c | 0.06 ± 0.00 c | 0.463 ± 0.01 b |
| Sample | Ton-set (°C) | Td (°C) | ΔT(1/2) | ΔH (J·g−1) |
|---|---|---|---|---|
| NP | 58.89 ± 0.48 c | 66.62 ± 0.61 a | 5.84 ± 0.98 b,c | 24.03 ± 1.93 a |
| 250 MPa | 60.06 ± 0.27 a | 65.98 ± 0.28 b,c | 5.32 ± 1.98 d | 12.56 ± 2.32 b |
| 350 MPa | 58.53 ± 0.32 c | 65.88 ± 0.37 b | 5.56 ± 0.76 c | 6.97 ± 1.53 c |
| 450 MPa | 59.99 ± 0.81 b | 65.88 ± 0.54 b | 7.07 ± 0.58 a | 5.72 ± 2.06 d |
| 550 MPa | 60.41 ± 0.42 a | 65.64 ± 0.31 b,c | 6.04 ± 0.98 b | 3.05 ± 0.32 e |
| Sample | α-Helix | β-Strand | β-Turn | Random Coil |
|---|---|---|---|---|
| NP | 24.2 a | 24.2 e | 21.2 a | 30.3 d |
| 250 MPa | 21.7 b | 26.5 d | 21.0 a | 30.9 c,d |
| 350 MPa | 16.3 c | 31.6 c | 20.6 b | 31.4 c |
| 450 MPa | 7.3 d | 36.4 b | 21.4 a | 34.8 b |
| 550 MPa | 4.1 e | 39.5 a | 19.2 c | 37.2 a |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elahi, R.; Mu, T.-H. High Hydrostatic Pressure (HHP)-Induced Structural Modification of Patatin and Its Antioxidant Activities. Molecules 2017, 22, 438. https://doi.org/10.3390/molecules22030438
Elahi R, Mu T-H. High Hydrostatic Pressure (HHP)-Induced Structural Modification of Patatin and Its Antioxidant Activities. Molecules. 2017; 22(3):438. https://doi.org/10.3390/molecules22030438
Chicago/Turabian StyleElahi, Rizwan, and Tai-Hua Mu. 2017. "High Hydrostatic Pressure (HHP)-Induced Structural Modification of Patatin and Its Antioxidant Activities" Molecules 22, no. 3: 438. https://doi.org/10.3390/molecules22030438
APA StyleElahi, R., & Mu, T. -H. (2017). High Hydrostatic Pressure (HHP)-Induced Structural Modification of Patatin and Its Antioxidant Activities. Molecules, 22(3), 438. https://doi.org/10.3390/molecules22030438