
Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin

Abstract
:1. Introduction
2. Results
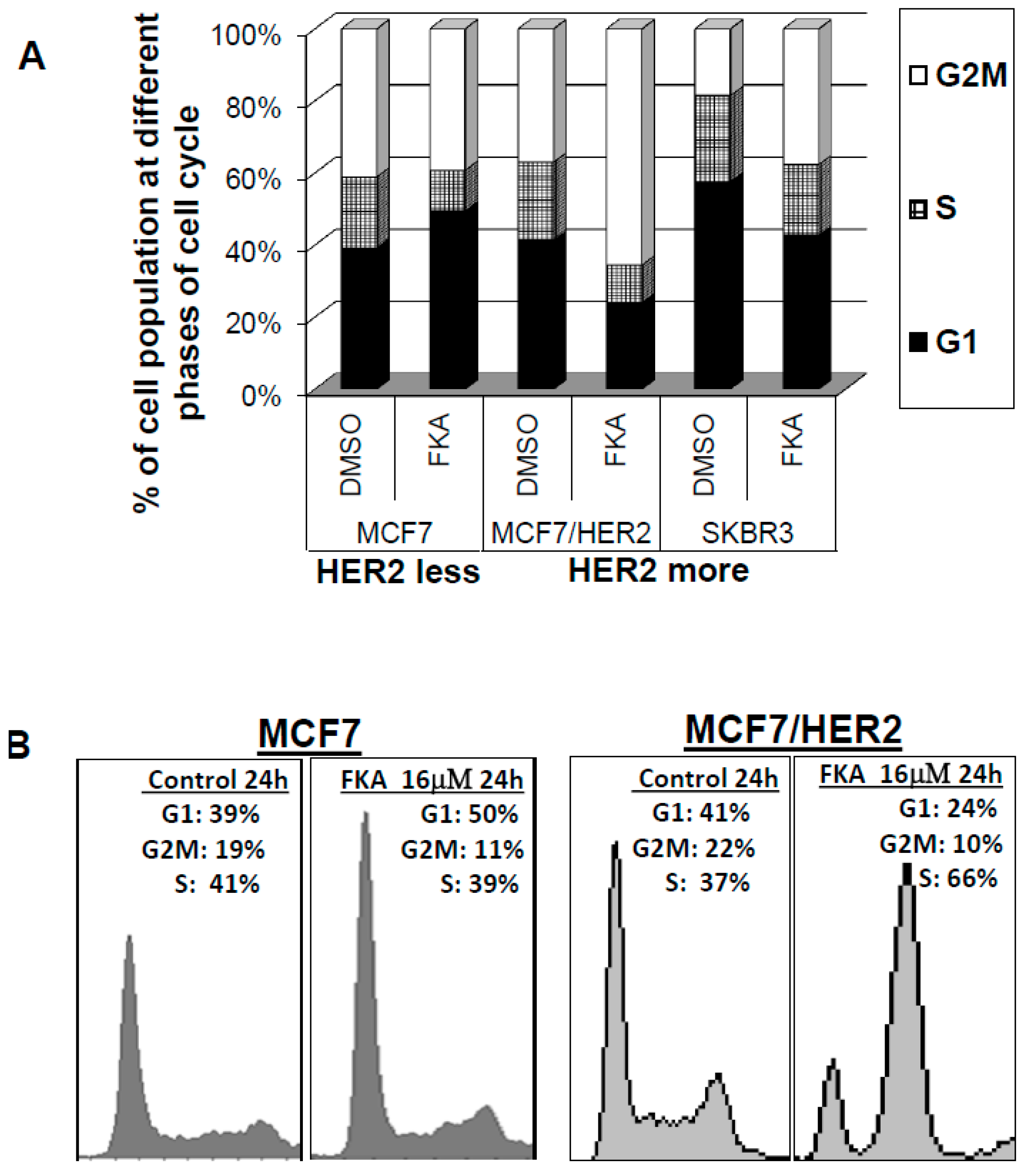
2.1. The Effect of FKA on Cell Cycle Progression Differs between HER2 Overexpressing versus Low-Expressing Breast Cancer Cell Lines
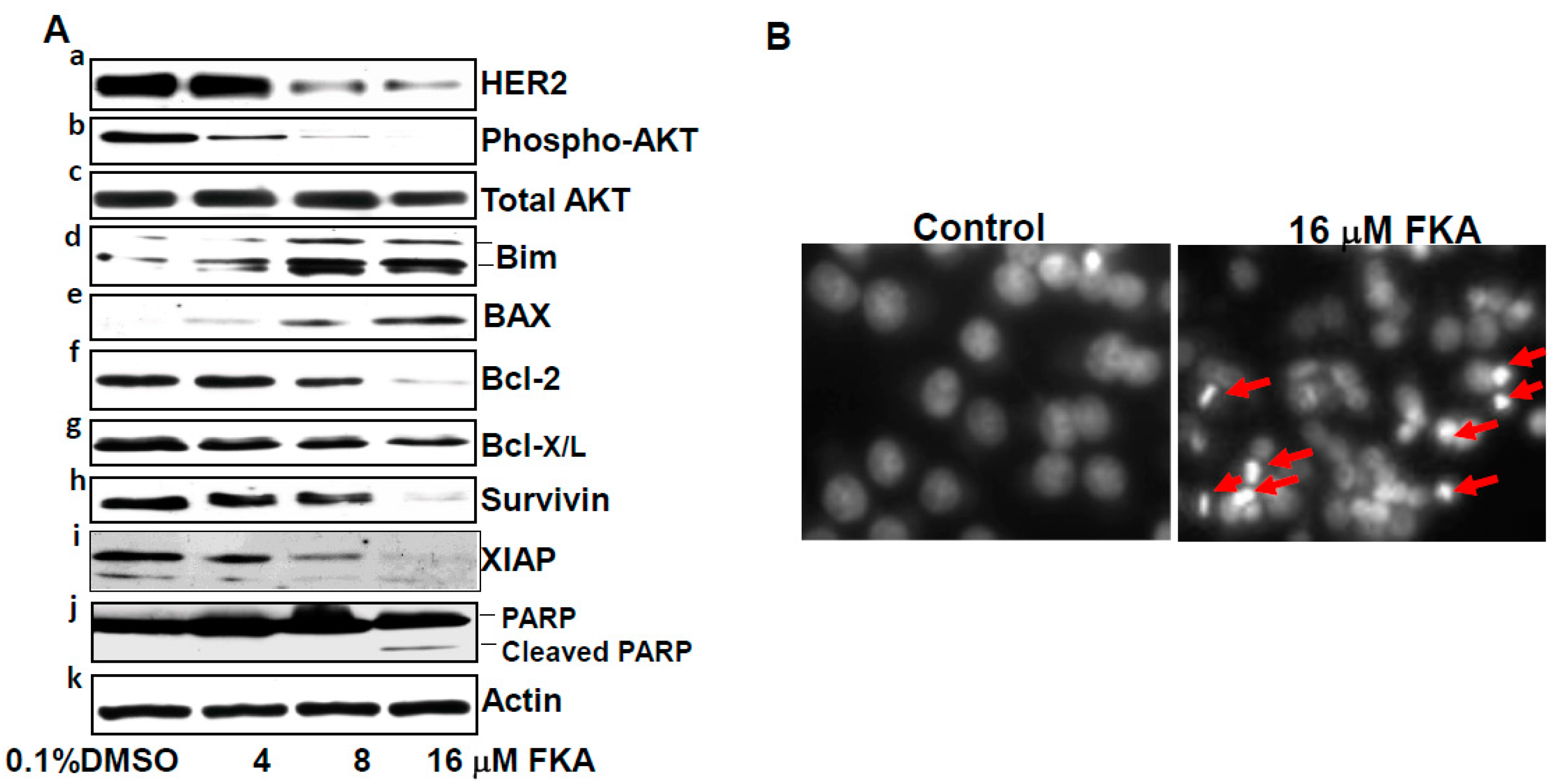
2.2. The Mechanisms of FK A–Induced G2M Arrest in HER2-Overexpressing SKBR3 Cells Are Associated with Inhibition of Cdc2 Phosphorylation via Downregulation of Wyt1 and Wee1 Expression and Cdc25C Phosphorylation
2.3. FKA Induces Apoptosis in HER2-Overexpressing Breast Cancer SKBR3 Cells
2.4. Herceptin and FKA Combination Causes Enhanced Growth Inhibitory Effect on HER2-Overexpressing Breast Cancer Cells via Down-Regulation of Myt1, Wee1, Survivin, and XIAP Expression
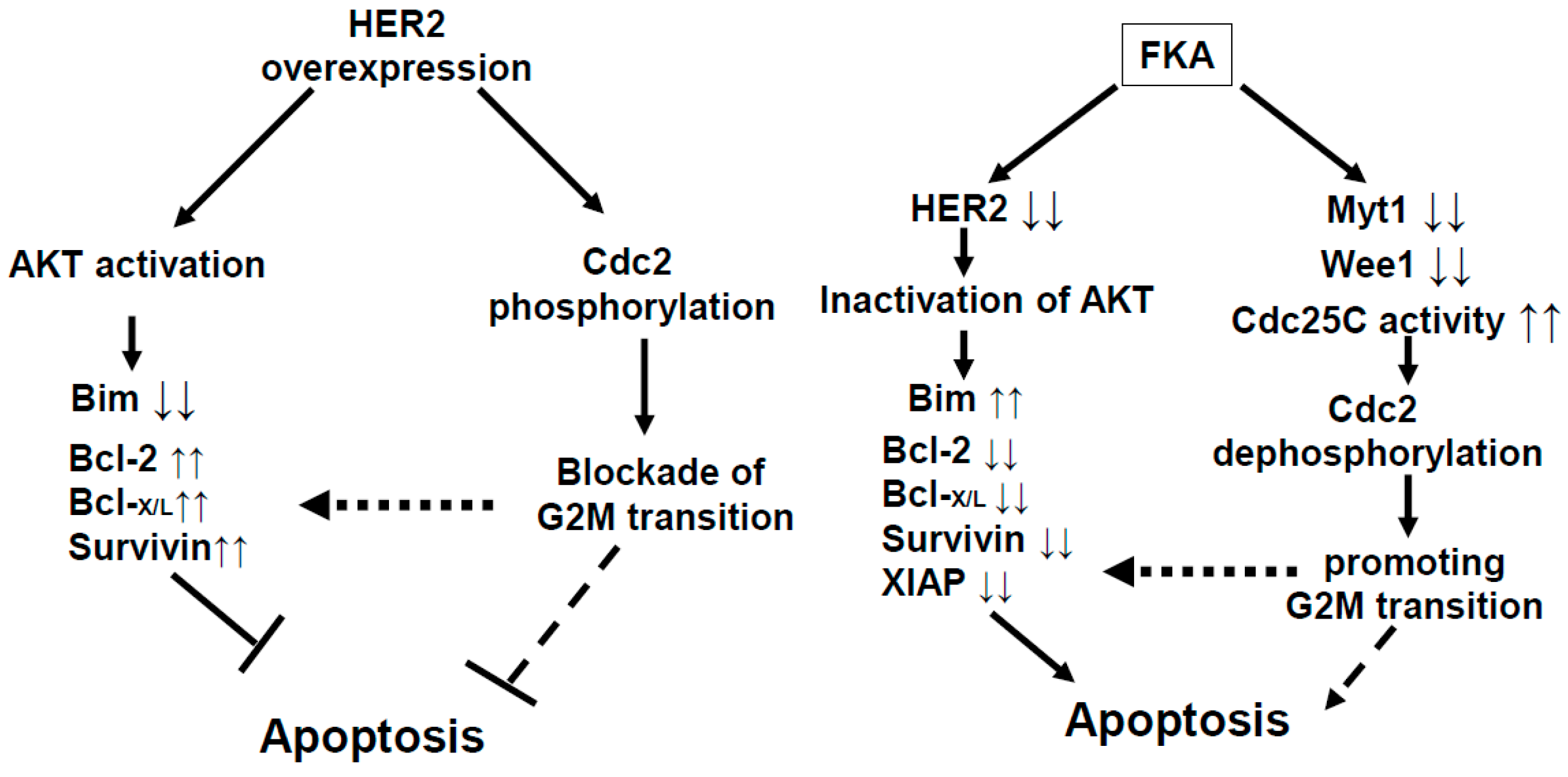
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Compounds, and Reagents
4.2. MTT Assay
4.3. Soft Agar Colony Formation
4.4. Flow Cytometric Analysis of Cell Cycle Distribution
4.5. Western Blotting Analysis
4.6. In Vitro Kinase Assay
4.7. 4',6-Diamidino-2-phenylindole (DAPI) Nuclear Staining
5. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hurvitz, S.A.; Lalla, D.; Guerin, A.; Latremouille-Viau, D.; Yu, A.P.; Wu, E.Q.; Brammer, M. Chemotherapy-related complication burden in patients with metastatic breast cancer in a real-world setting. J. Clin. Oncol. 2011, 29, e11101. [Google Scholar] [CrossRef]
- Sodergren, S.C.; Copson, E.; White, A.; Efficace, F.; Sprangers, M.; Fitzsimmons, D.; Bottomley, A.; Johnson, C.D. Systematic Review of the Side Effects Associated With Anti-HER2-Targeted Therapies Used in the Treatment of Breast Cancer, on Behalf of the EORTC Quality of Life Group. Targeted Oncol. 2016, 11, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Bacus, S.S.; Gudkov, A.V.; Esteva, F.J.; Yarden, Y. Expression of erbB receptors and their ligands in breast cancer: Implications to biological behavior and therapeutic response. Breast Dis. 2000, 11, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Ariazi, J.L.; Cordera, F.; Jordan, V.C. Estrogen receptors as therapeutic targets in breast cancer. Curr. Top. Med. Chem. 2006, 6, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N. Overview of treatment results with trastuzumab (Herceptin) in metastatic breast cancer. Semin. Oncol. 2001, 28, 43–47. [Google Scholar] [CrossRef]
- Smith, K.L.; Dang, C.; Seidman, A.D. Cardiac dysfunction associated with trastuzumab. Expert Opin. Drug Saf. 2006, 5, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Siziopikou, K.P.; Khan, S. Correlation of HER2 gene amplification with expression of the apoptosis-suppressing genes bcl-2 and bcl-x-L in ductal carcinoma in situ of the breast. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Thor, A.D.; Liu, S.; Edgerton, S.; Moore, D., 2nd; Kasowitz, K.M.; Benz, C.C.; Stern, D.F.; DiGiovanna, M.P. Activation (tyrosine phosphorylation) of ErbB-2 (HER-2/neu): A study of incidence and correlation with outcome in breast cancer. J. Clin. Oncol. 2000, 18, 3230–3239. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Lloveras, B.; Figueras, A.; Escobedo, A.; Ramon, J.M.; Sierra, A.; Fabra, A. Ductal carcinoma in situ of the breast: Correlation between histologic classification and biologic markers. Mod. Pathol. 1997, 10, 1088–1092. [Google Scholar] [PubMed]
- Bartkova, J.; Barnes, D.M.; Millis, R.R.; Gullick, W.J. Immunohistochemical demonstration of c-erbB-2 protein in mammary ductal carcinoma in situ. Hum. Pathol. 1990, 21, 1164–1167. [Google Scholar] [CrossRef]
- De Placido, S.; Carlomagno, C.; De Laurentiis, M.; Bianco, A.R. c-erbB2 expression predicts tamoxifen efficacy in breast cancer patients. Breast Cancer Res. Treat. 1998, 52, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Zi, X.; Simoneau, A.R. Flavokawain A, a novel chalcone from kava extract, induces apoptosis in bladder cancer cells by involvement of Bax protein-dependent and mitochondria-dependent apoptotic pathway and suppresses tumor growth in mice. Cancer Res. 2005, 65, 3479–3486. [Google Scholar] [PubMed]
- Tang, Y.; Simoneau, A.R.; Xie, J.; Shahandeh, B.; Zi, X. Effects of the kava chalcone flavokawain A differ in bladder cancer cells with wild-type versus mutant p53. Cancer Prev. Res. 2008, 1, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Abu, N.; Akhtar, M.N.; Yeap, S.K.; Lim, K.L.; Ho, W.Y.; Zulfadli, A.J.; Omar, A.R.; Sulaiman, M.R.; Abdullah, M.P.; Alitheen, N.B. Flavokawain A induces apoptosis in MCF-7 and MDA-MB231 and inhibits the metastatic process in vitro. PLoS ONE 2014, 9, e105244. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yokoyama, N.N.; Zhang, S.; Ding, L.; Liu, H.M.; Lilly, M.B.; Mercola, D.; Zi, X. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget 2015, 6, 41809–41824. [Google Scholar] [PubMed]
- Abu, N.; Mohamed, N.E.; Yeap, S.K.; Lim, K.L.; Akhtar, M.N.; Zulfadli, A.J.; Kee, B.B.; Abdullah, M.P.; Omar, A.R.; Alitheen, N.B. In Vivo Anti-Tumor Effects of Flavokawain A in 4T1 Breast Cancer Cell-Challenged Mice. Anticancer Agents Med. Chem. 2015, 15, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, X.; Li, X.; Liu, S.; Simoneau, A.R.; He, F.; Wu, X.R.; Zi, X. Kava chalcone, flavokawain A, inhibits urothelial tumorigenesis in the UPII-SV40T transgenic mouse model. Cancer Prev. Res. 2013, 6, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, X.; Ji, T.; Liu, Z.; Gu, M.; Hoang, B.H.; Zi, X. Dietary feeding of Flavokawain A, a Kava chalcone, exhibits a satisfactory safety profile and its association with enhancement of phase II enzymes in mice. Toxicol. Rep. 2014, 1, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Abu, N.; Mohameda, N.E.; Tangarajoo, N.; Yeap, S.K.; Akhtar, M.N.; Abdullah, M.P.; Omar, A.R.; Alitheen, N.B. In vitro Toxicity and in vivo Immunomodulatory Effects of Flavokawain A and Flavokawain B in Balb/C Mice. Nat. Prod. Commun. 2015, 10, 1199–1202. [Google Scholar] [PubMed]
- Van de Vijver, M.J.; Mooi, W.J.; Wisman, P.; Peterse, J.L.; Nusse, R. Immunohistochemical detection of the neu protein in tissue sections of human breast tumors with amplified neu DNA. Oncogene 1988, 2, 175–178. [Google Scholar] [PubMed]
- Miller, F.R.; Bukowski, J. Mammary tumor stimulatory factors as well as mammastatin are produced by the normal human breast epithelial cell line MCF10A. Int. J. Cancer 1994, 59, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Klos, K.S.; Zhou, X.; Yao, J.; Yang, Y.; Smith, T.L.; Shi, D.; Yu, D. ErbB2 overexpression in human breast carcinoma is correlated with p21Cip1 up-regulation and tyrosine-15 hyperphosphorylation of p34Cdc2: Poor responsiveness to chemotherapy with cyclophoshamide methotrexate, and 5-fluorouracil is associated with Erb2 overexpression and with p21Cip1 overexpression. Cancer 2003, 98, 1123–1130. [Google Scholar] [PubMed]
- Tapia, C.; Kutzner, H.; Mentzel, T.; Savic, S.; Baumhoer, D.; Glatz, K. Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. Am. J. Surg. Pathol. 2006, 30, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Henson, E.S.; Hu, X.; Gibson, S.B. Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin. Cancer Res. 2006, 12, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, H.; Torigoe, T.; Kamiguchi, K.; Hirohashi, Y.; Ohmura, T.; Hirata, K.; Sato, M.; Sato, N. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005, 65, 11018–11025. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mandal, M.; Lipton, A.; Harvey, H.; Thompson, C.B. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin. Cancer Res. 1996, 2, 1215–1219. [Google Scholar] [PubMed]
- Reginato, M.J.; Mills, K.R.; Becker, E.B.; Lynch, D.K.; Bonni, A.; Muthuswamy, S.K.; Brugge, J.S. Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol. Cell. Biol. 2005, 25, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Sandhu, N.; Herrmann, J. Systems biology approaches to adverse drug effects: The example of cardio-oncology. Nat. Rev. Clin. Oncol. 2015, 12, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Roulot, A.; Héquet, D.; Guinebretière, J.M.; Vincent-Salomon, A.; Lerebours, F.; Dubot, C.; Rouzier, R. Tumoral heterogeneity of breast cancer. Ann. Biol. Clin. 2016, 74, 653–660. [Google Scholar]
- Choudhury, A.; Kiessling, R. Her-2/neu as a paradigm of a tumor-specific target for therapy. Breast Dis. 2004, 20, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 53, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Guthei, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Shou, J.; Massarweh, S.; Schiff, R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin. Cancer Res. 2005, 11, 865s–870s. [Google Scholar] [PubMed]
- Ju, J.H.; Yang, W.; Oh, S.; Nam, K.; Lee, K.M.; Noh, D.Y.; Shin, I. HER2 stabilizes survivin while concomitantly down-regulating survivin gene transcription by suppressing Notch cleavage. Biochem. J. 2013, 45, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.J.; Ju, S.M.; Youn, G.S.; Choi, S.Y.; Park, J. Suppression of iNOS and COX-2 expression by flavokawain A via blockade of NF-κB and AP-1 activation in RAW 264.7 macrophages. Food Chem. Toxicol. 2013, 58, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Hein, A.L.; Greer, P.M.; Wang, Z.; Kolb, R.H.; Batra, S.K.; Cowan, K.H. A novel function of HER2/Neu in the activation of G2/M checkpoint in response to γ-irradiation. Oncogene 2015, 34, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Jing, T.; Lan, K.H.; Neal, C.L.; Li, P.; Lee, S.; Fang, D.; Nagata, Y.; Liu, J.; Arlinghaus, R.; et al. Phosphorylation on tyrosine-15 of p34(Cdc2) by ErbB2 inhibits p34(Cdc2) activation and is involved in resistance to taxol-induced apoptosis. Mol. Cell 2002, 9, 993–1004. [Google Scholar] [CrossRef]
- Xu, D.; Li, C.F.; Zhang, X.; Gong, Z.; Chan, C.H.; Lee, S.W.; Jin, G.; Rezaeian, A.H.; Han, F.; Wang, J.; et al. Skp2-macroH2A1-CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat. Commun. 2015, 6, 6641. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef]
- Hu, R.; Aplin, A.E. Skp2 regulates G2/M progression in a p53-dependent manner. Mol. Biol. Cell 2008, 19, 4602–4610. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bauzon, F.; Ji, P.; Xu, X.; Sun, D.; Locker, J.; Sellers, R.S.; Nakayama, K.; Nakayama, K.I.; Cobrinik, D.; et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat. Genet. 2010, 42, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Chen, Z.; Wang, G.; Nardella, C.; Lee, S.W.; Chan, C.H.; Yang, W.L.; Wang, J.; Egia, A.; Nakayama, K.I.; et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010, 464, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Pollak, M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int. J. Cancer 2004, 108, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Li, C.F.; Yang, W.L.; Gao, Y.; Lee, S.W.; Feng, Z.; Huang, H.Y.; Tsai, K.K.; Flores, L.G.; Shao, Y.; et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012, 149, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds in small amounts are available from the authors.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCF10A | MDA-MB468 | MCF7 | MCF7/HER2 | SKBR3 | |
|---|---|---|---|---|---|
| Estrogen receptor | absent | mutant | wild-type | wild-type | mutant |
| P53 | wild-type | mutant | wild-type | wild-type | mutant |
| HER2 expression | + | + | + | ++++ | ++++ |
| IC50s | >100 μM | 38.4 μM | 45 μM | 13.6 μM | 10 μM |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandial, D.D.; Krill, L.S.; Chen, L.; Wu, C.; Ke, Y.; Xie, J.; Hoang, B.H.; Zi, X. Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin. Molecules 2017, 22, 462. https://doi.org/10.3390/molecules22030462
Jandial DD, Krill LS, Chen L, Wu C, Ke Y, Xie J, Hoang BH, Zi X. Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin. Molecules. 2017; 22(3):462. https://doi.org/10.3390/molecules22030462
Chicago/Turabian StyleJandial, Danielle D., Lauren S. Krill, Lixia Chen, Chunli Wu, Yu Ke, Jun Xie, Bang H. Hoang, and Xiaolin Zi. 2017. "Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin" Molecules 22, no. 3: 462. https://doi.org/10.3390/molecules22030462
APA StyleJandial, D. D., Krill, L. S., Chen, L., Wu, C., Ke, Y., Xie, J., Hoang, B. H., & Zi, X. (2017). Induction of G2M Arrest by Flavokawain A, a Kava Chalcone, Increases the Responsiveness of HER2-Overexpressing Breast Cancer Cells to Herceptin. Molecules, 22(3), 462. https://doi.org/10.3390/molecules22030462