
Synthesis of Substituted α-Trifluoromethyl Piperidinic Derivatives


Abstract
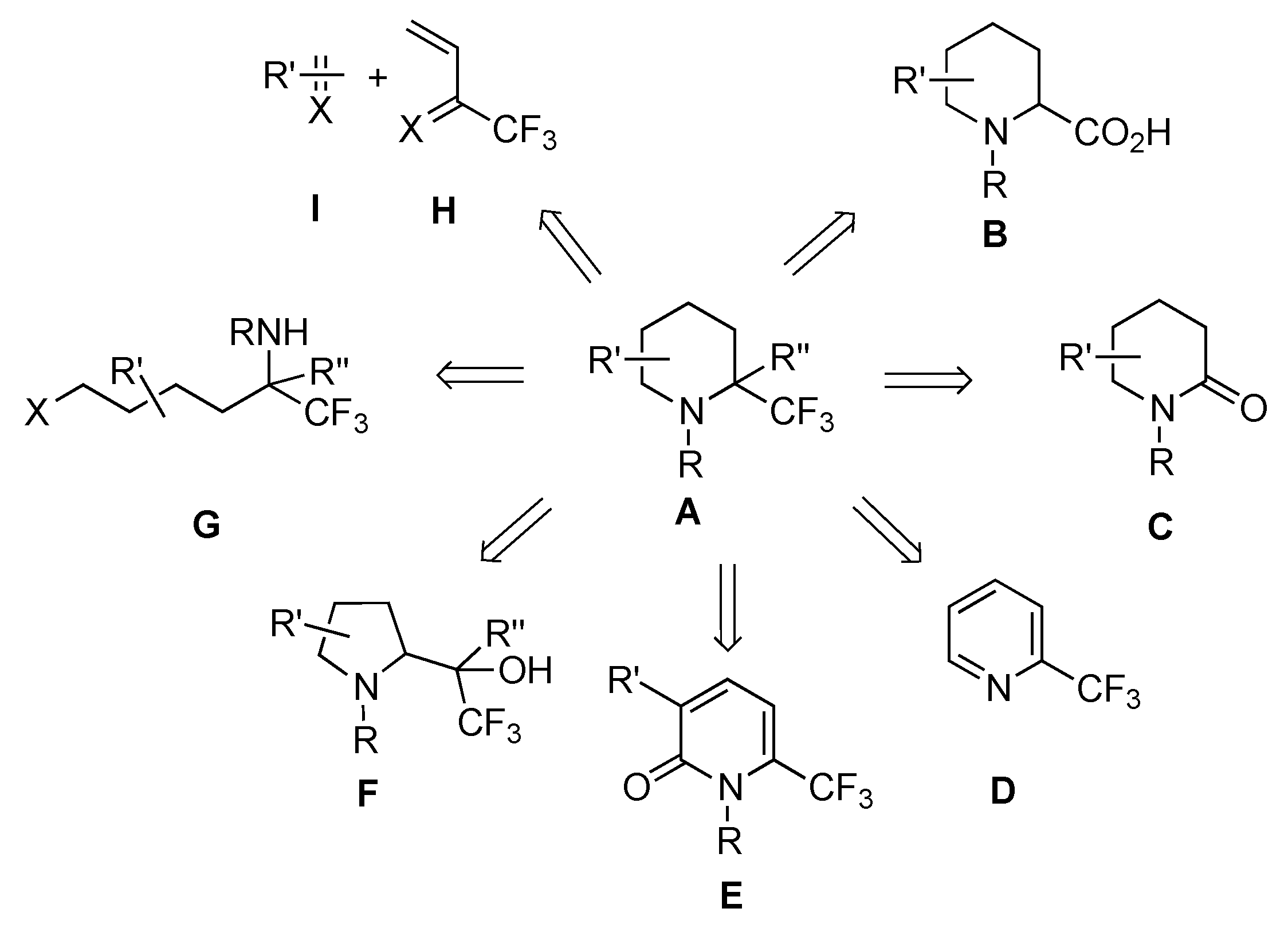
:1. Introduction
2. From Cyclic Substrates
2.1. From 6-Membered Rings
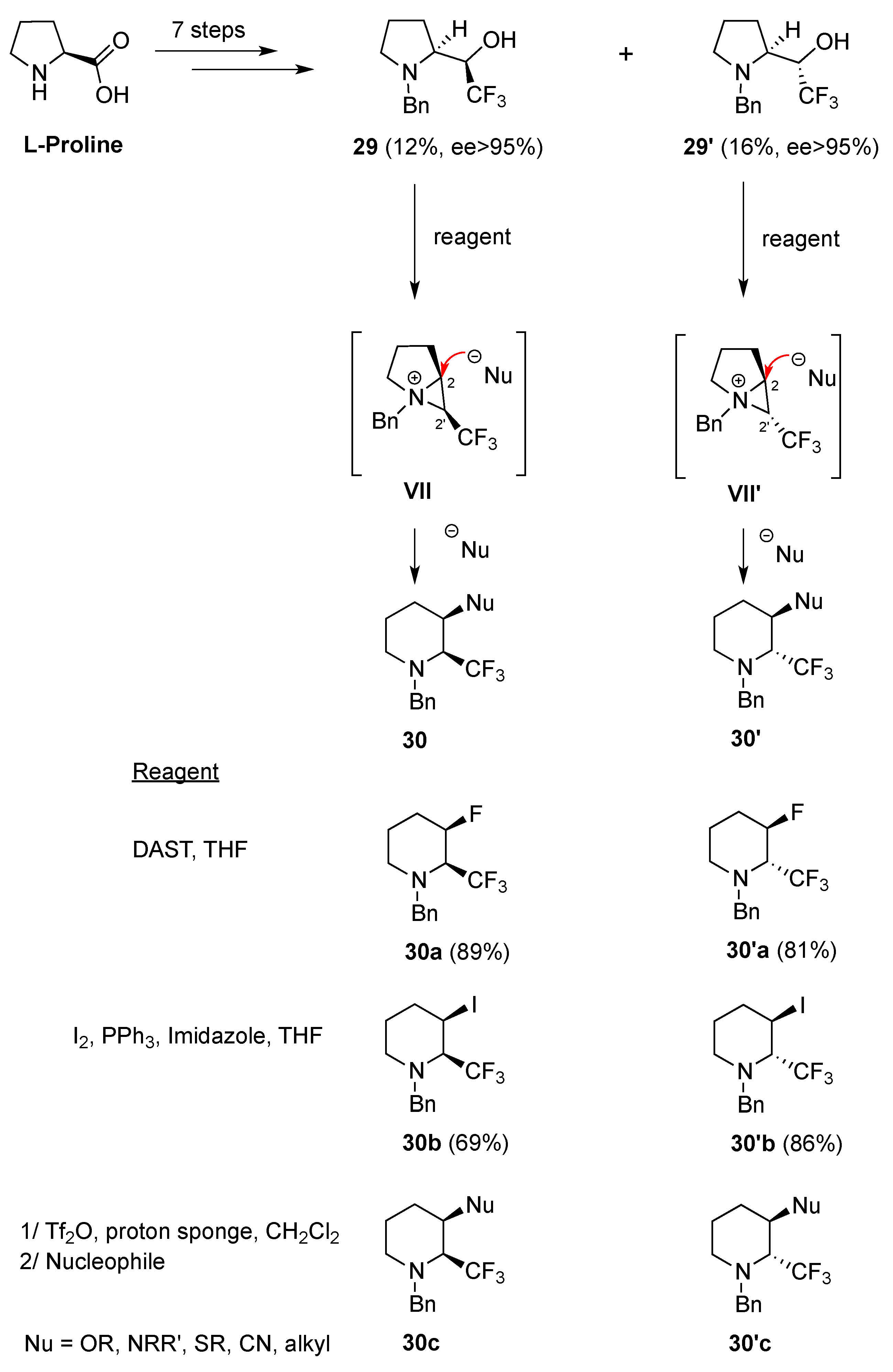
2.1.1. From Pipecolic Acid
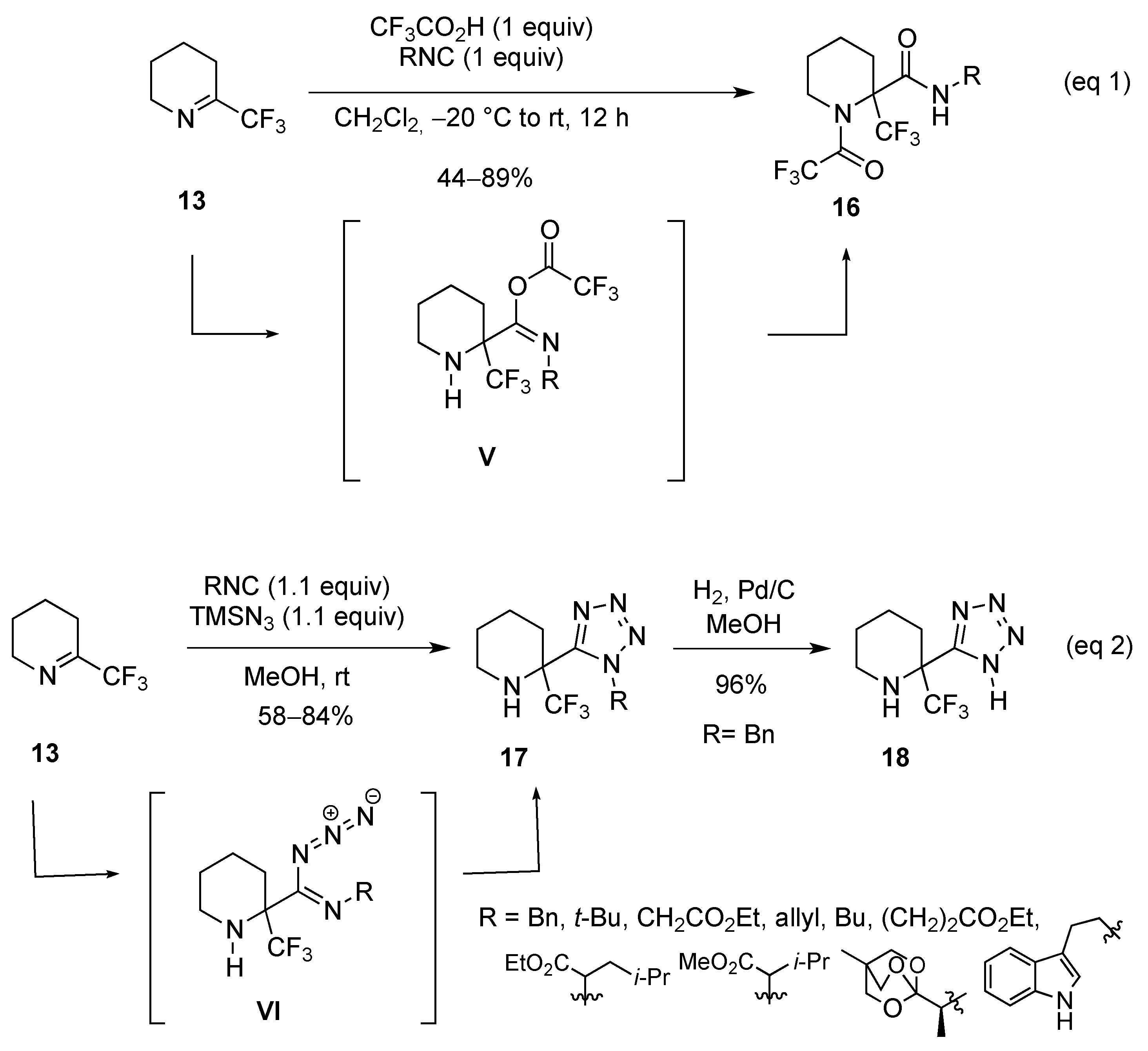
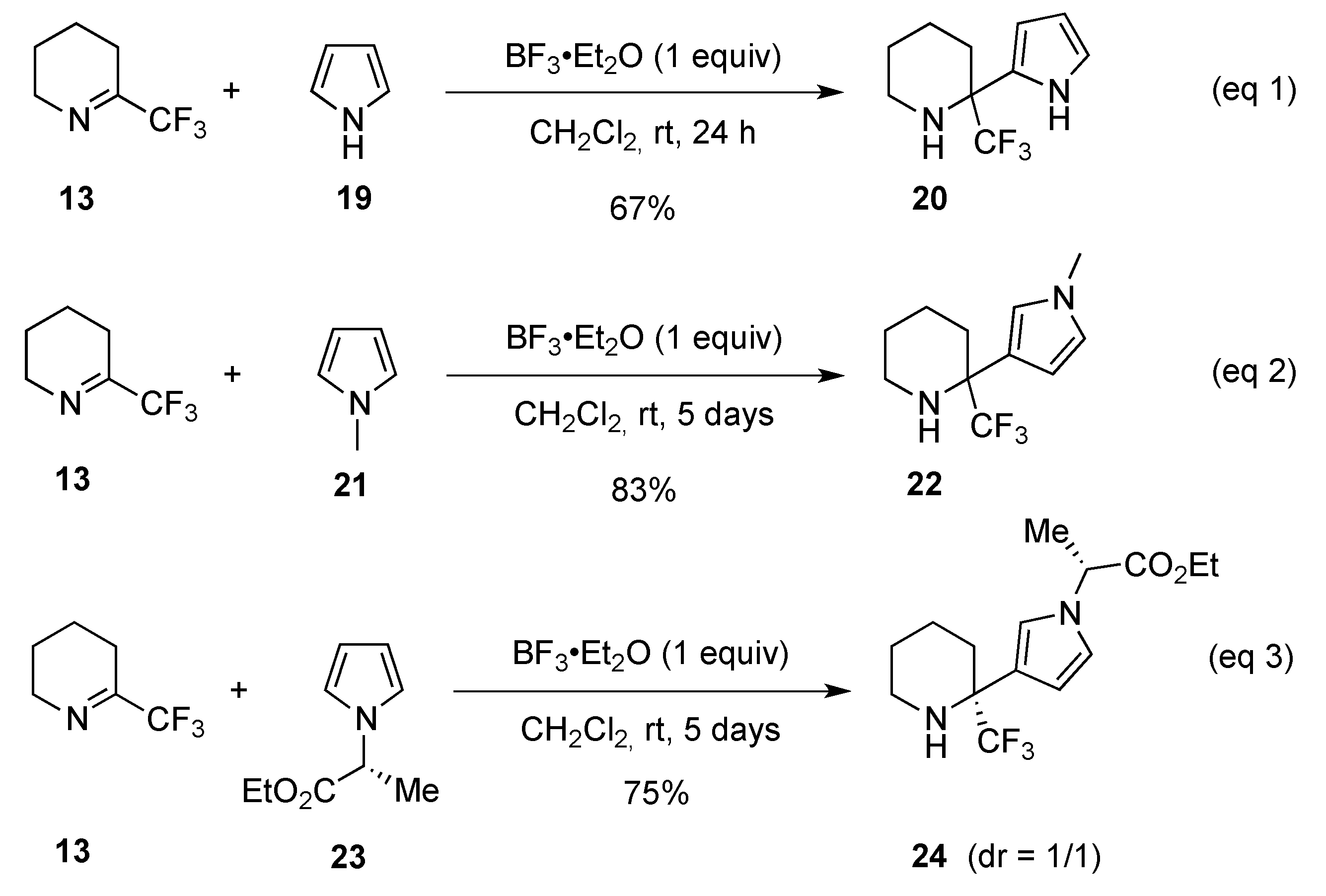
2.1.2. From 2-Trifluoromethylpyridine
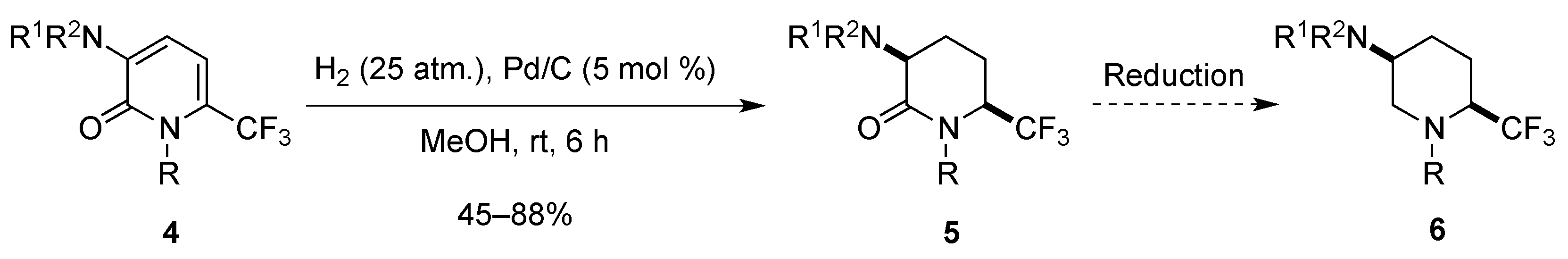
2.1.3. From Trifluoromethyl Pyridinones
2.1.4. From δ-Lactams
From Enamines C1
From Imines C2
From Imines C3
2.2. From 5-Membered Rings
3. From Non-cyclic Substrates
3.1. Cyclization
3.1.1. By Metathesis
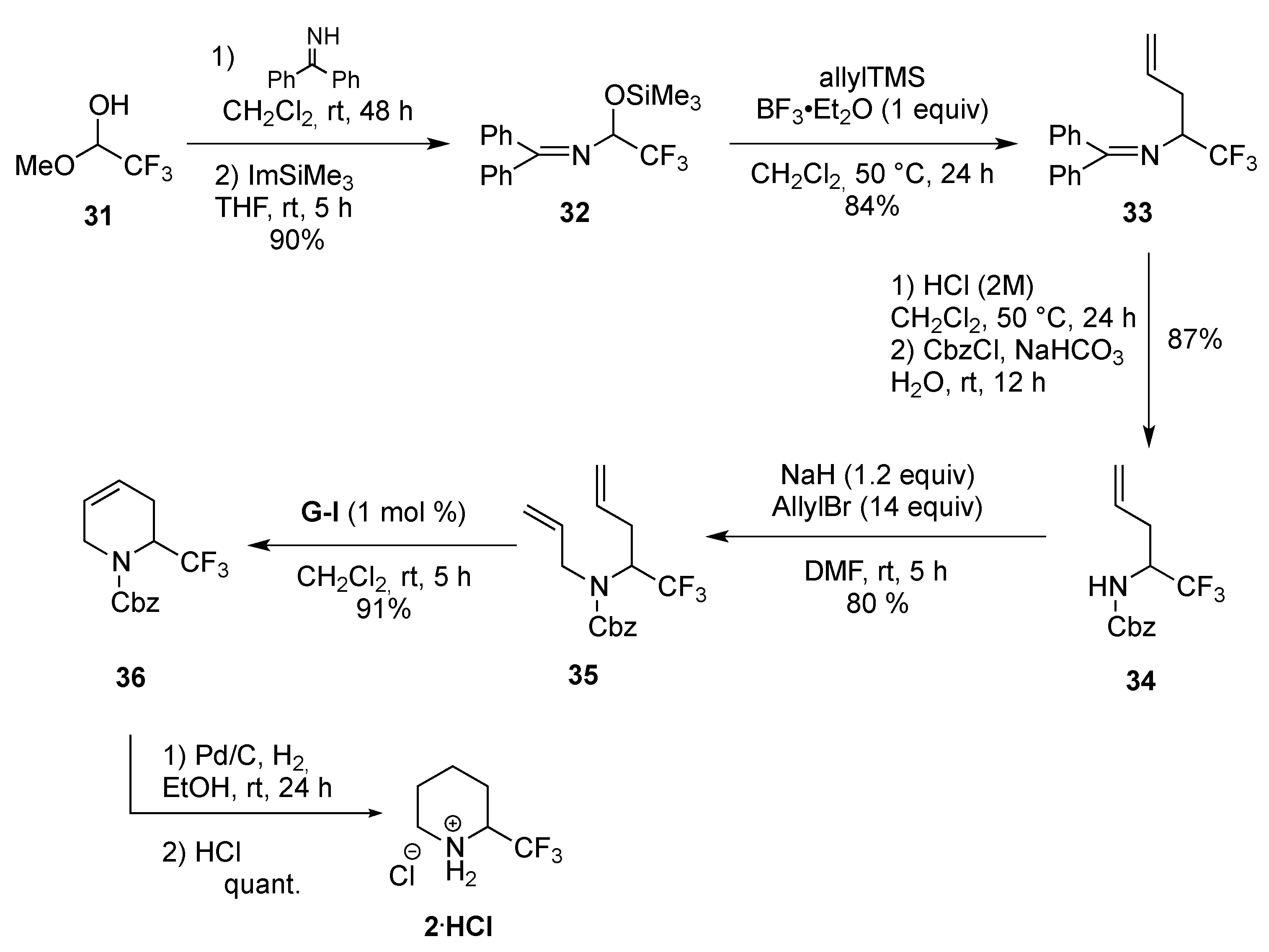
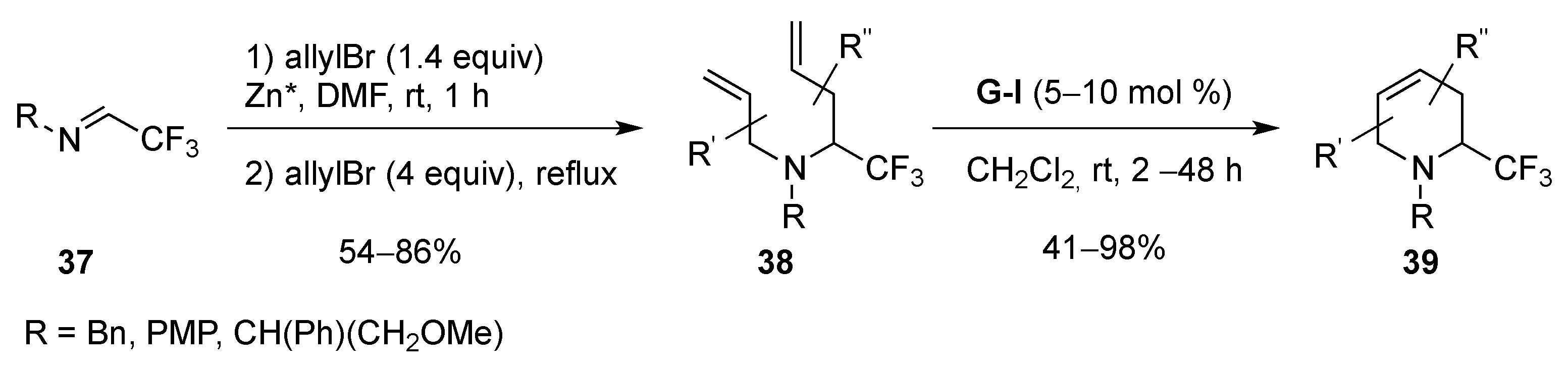
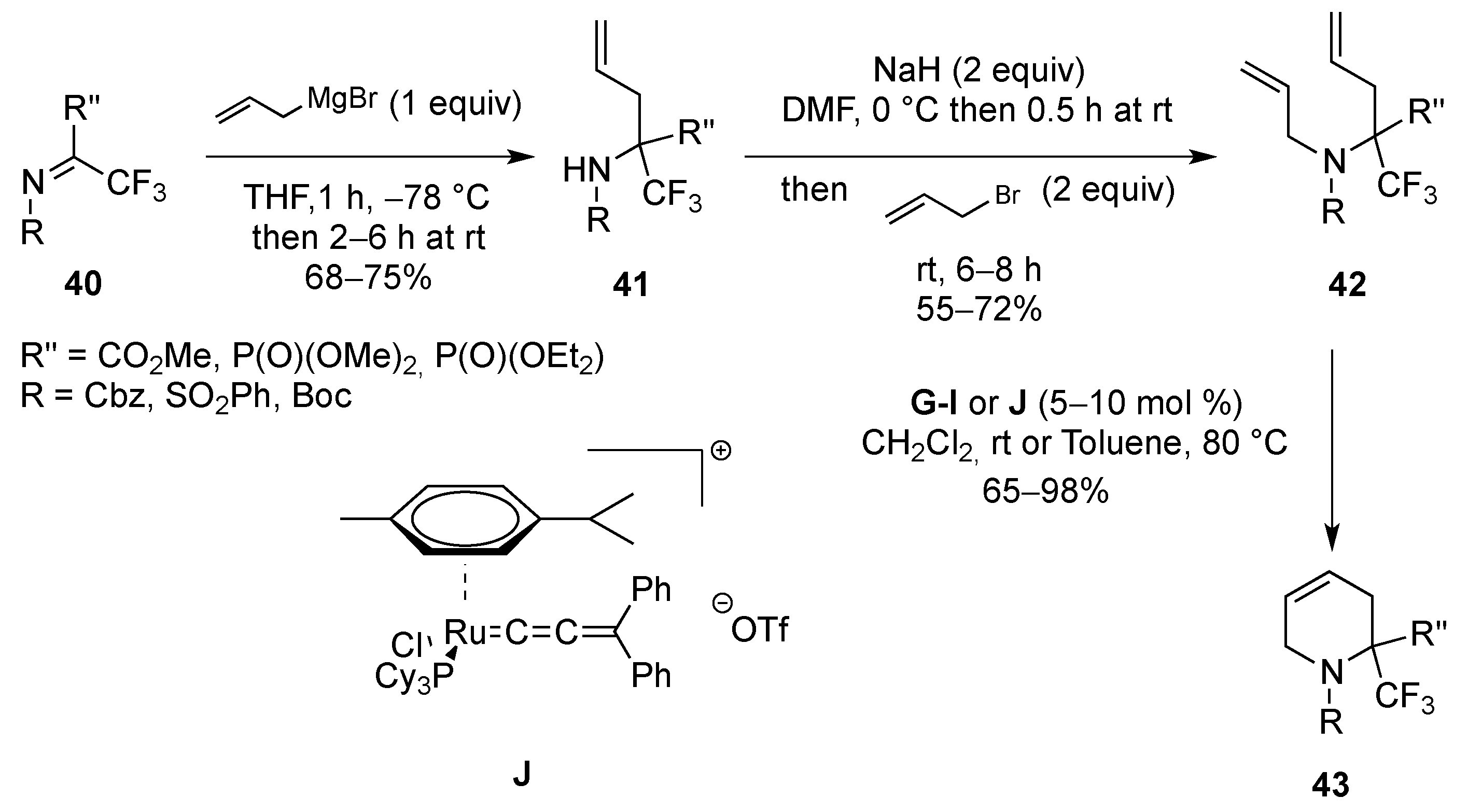
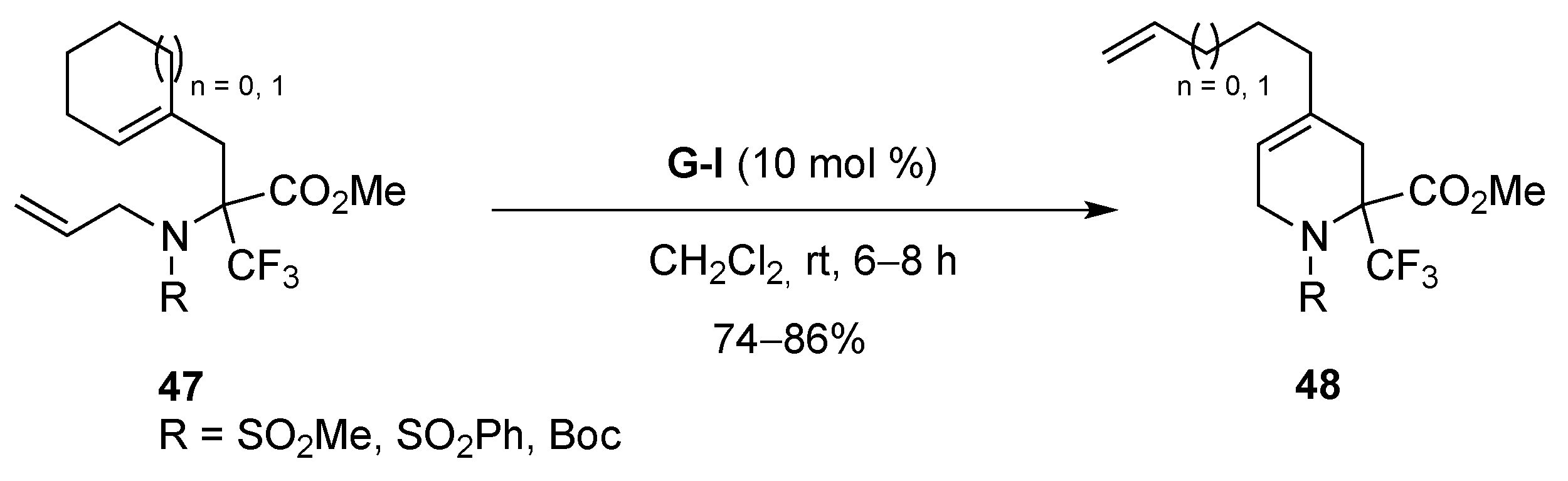
From Dienes
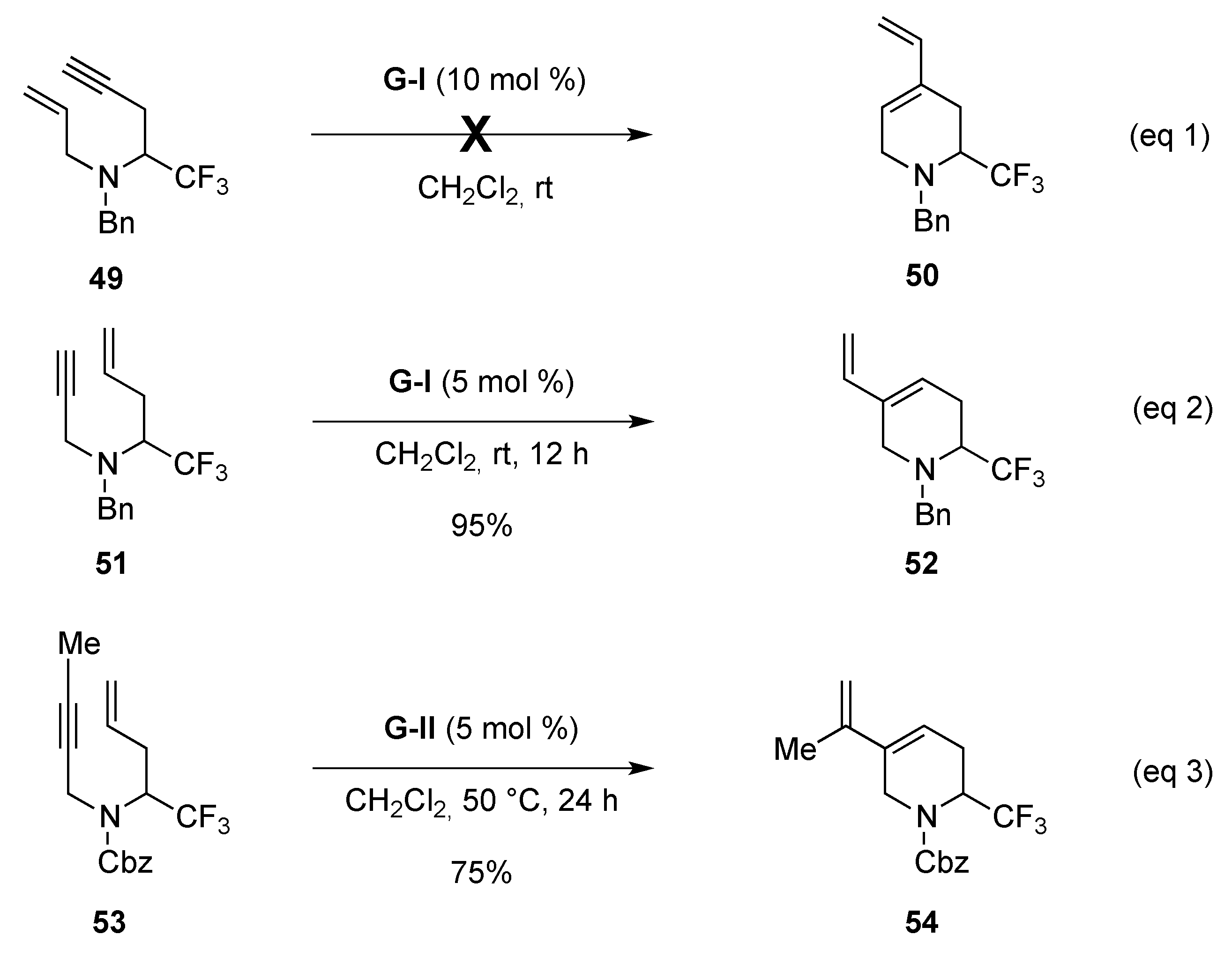
From Enynes
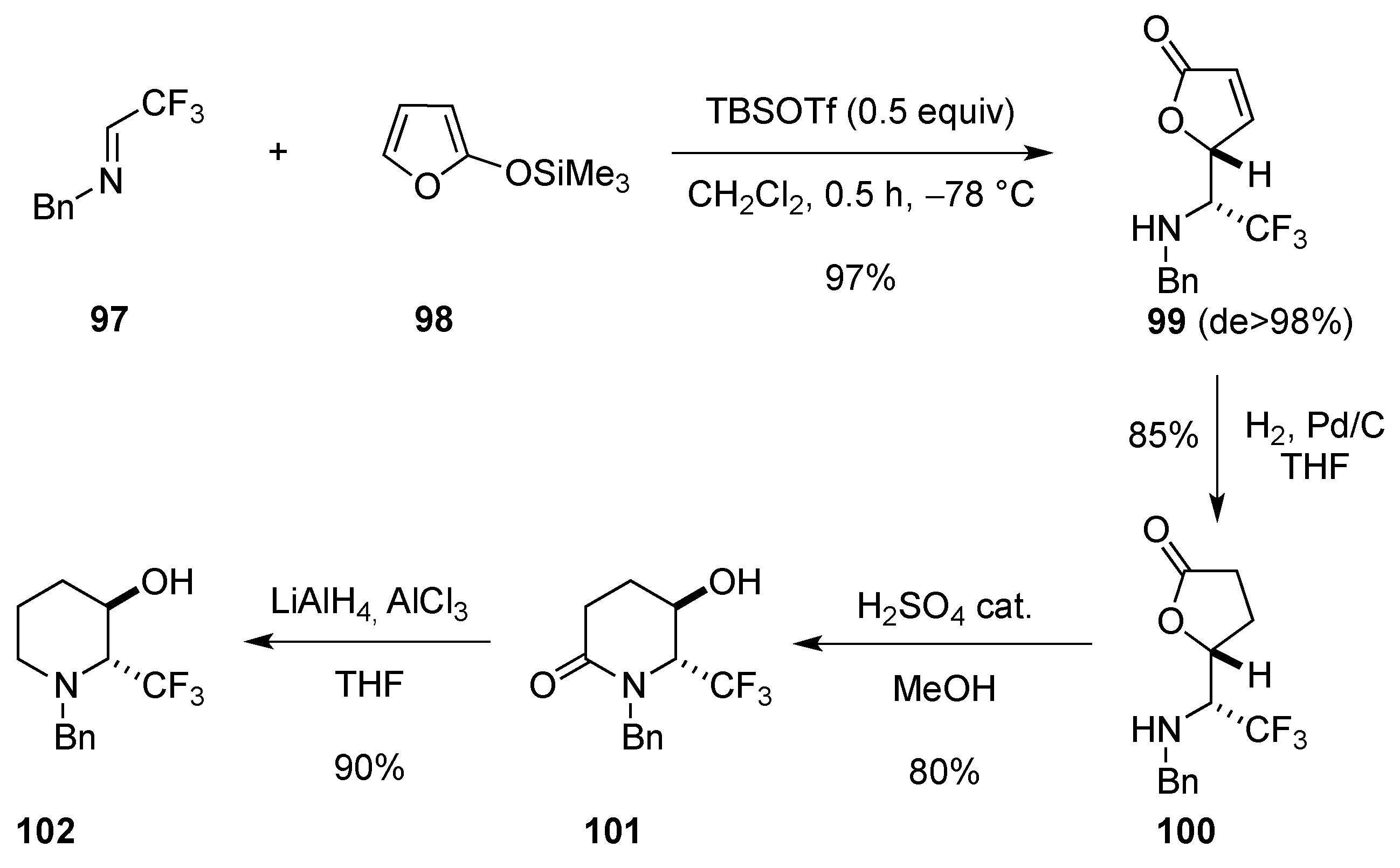
3.1.2. By Cycloaddition
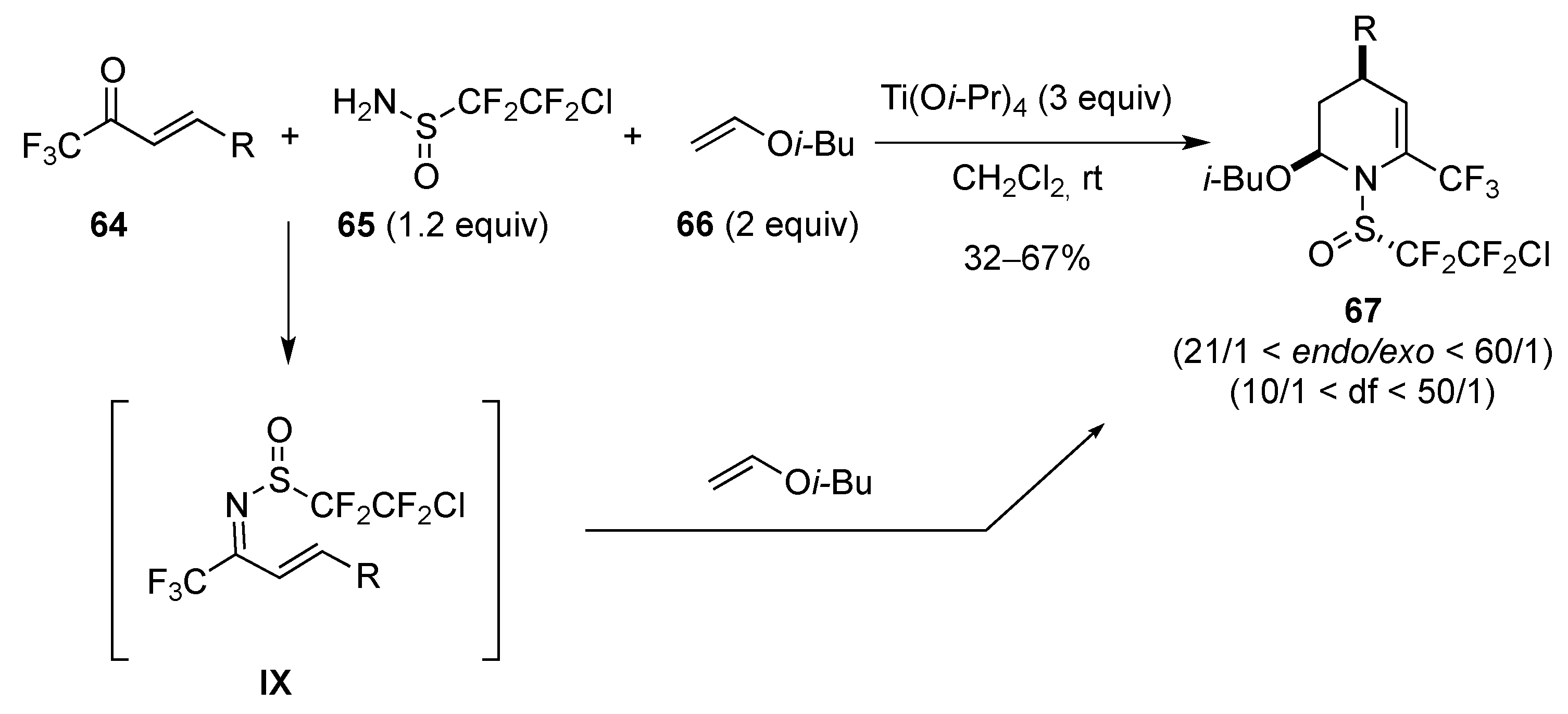
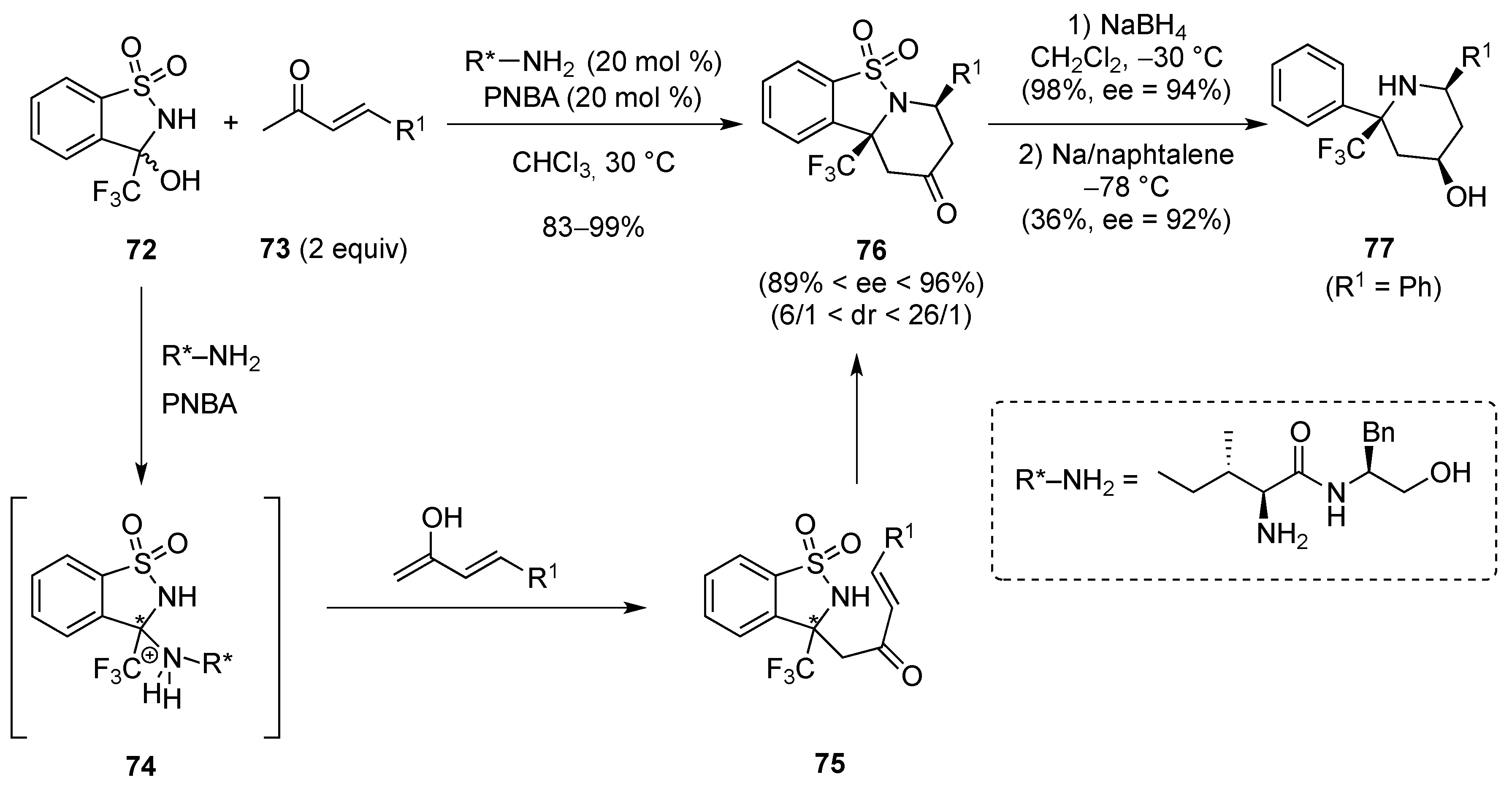
[4 + 2]-Cycloaddition: Aza Diels-Alder
Aza Diels-Alder Reaction with 1-Azadiene
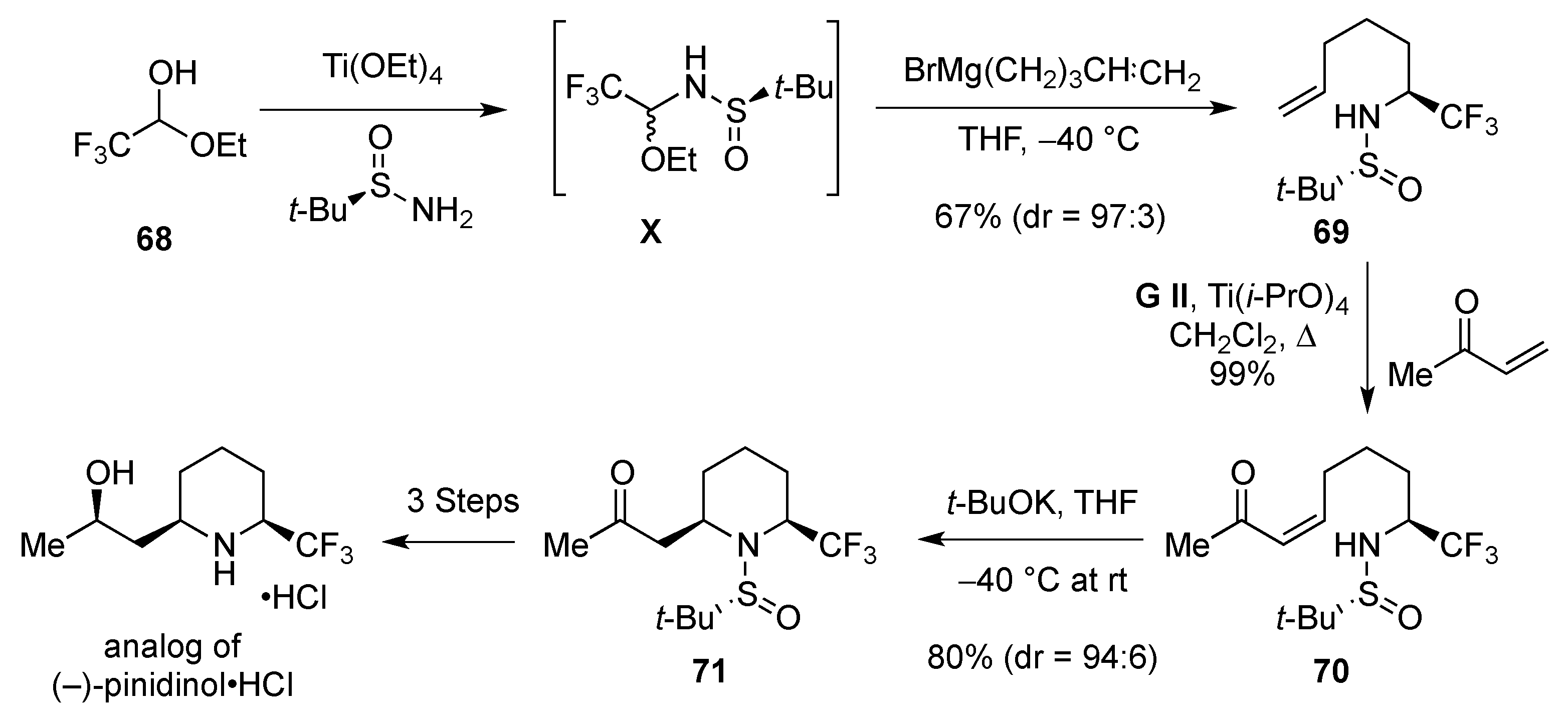
3.1.3. 1,4-Addition: Aza-Michael
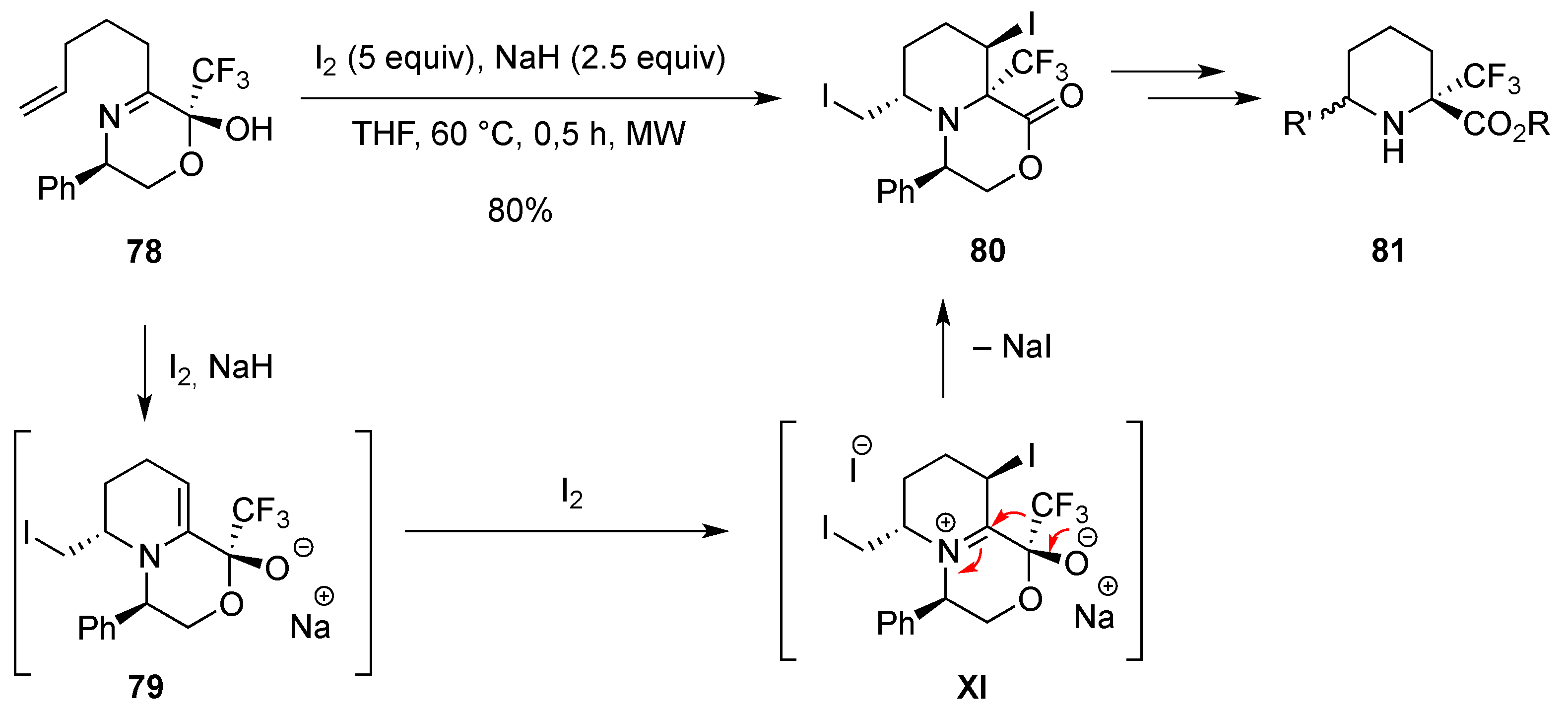
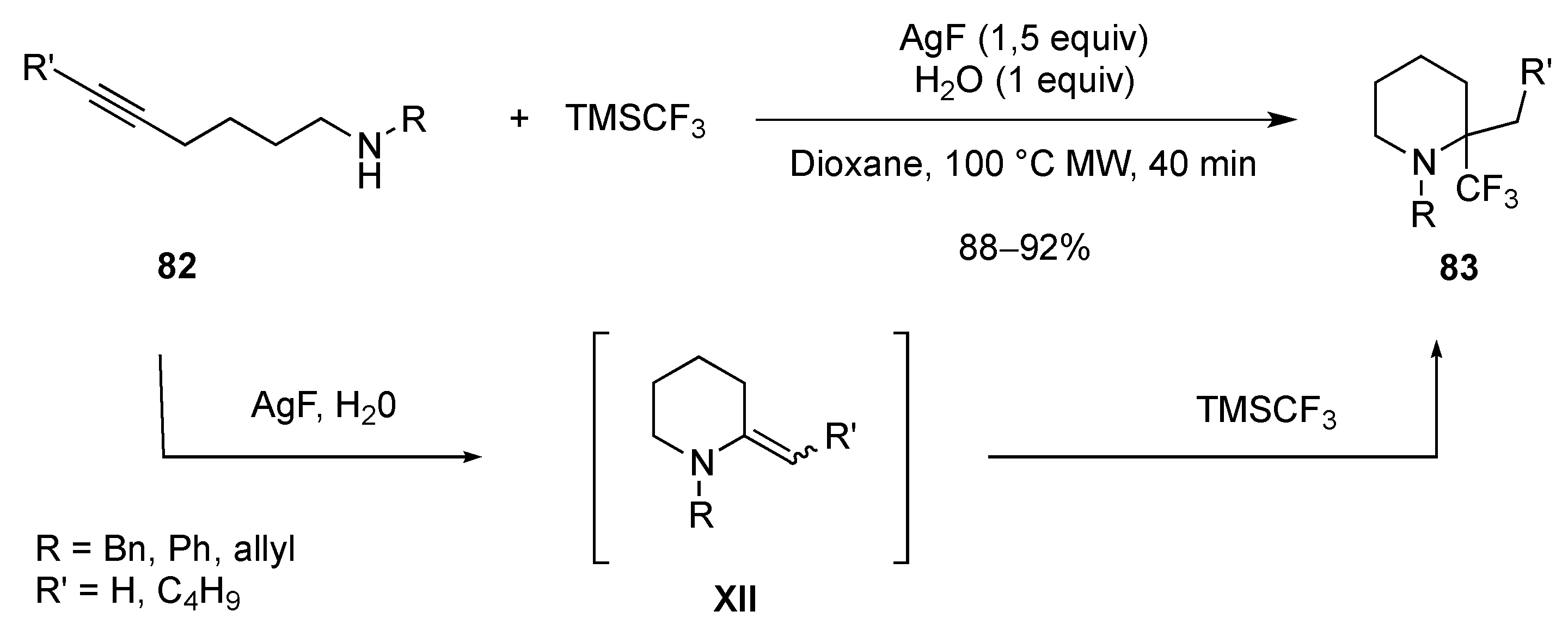
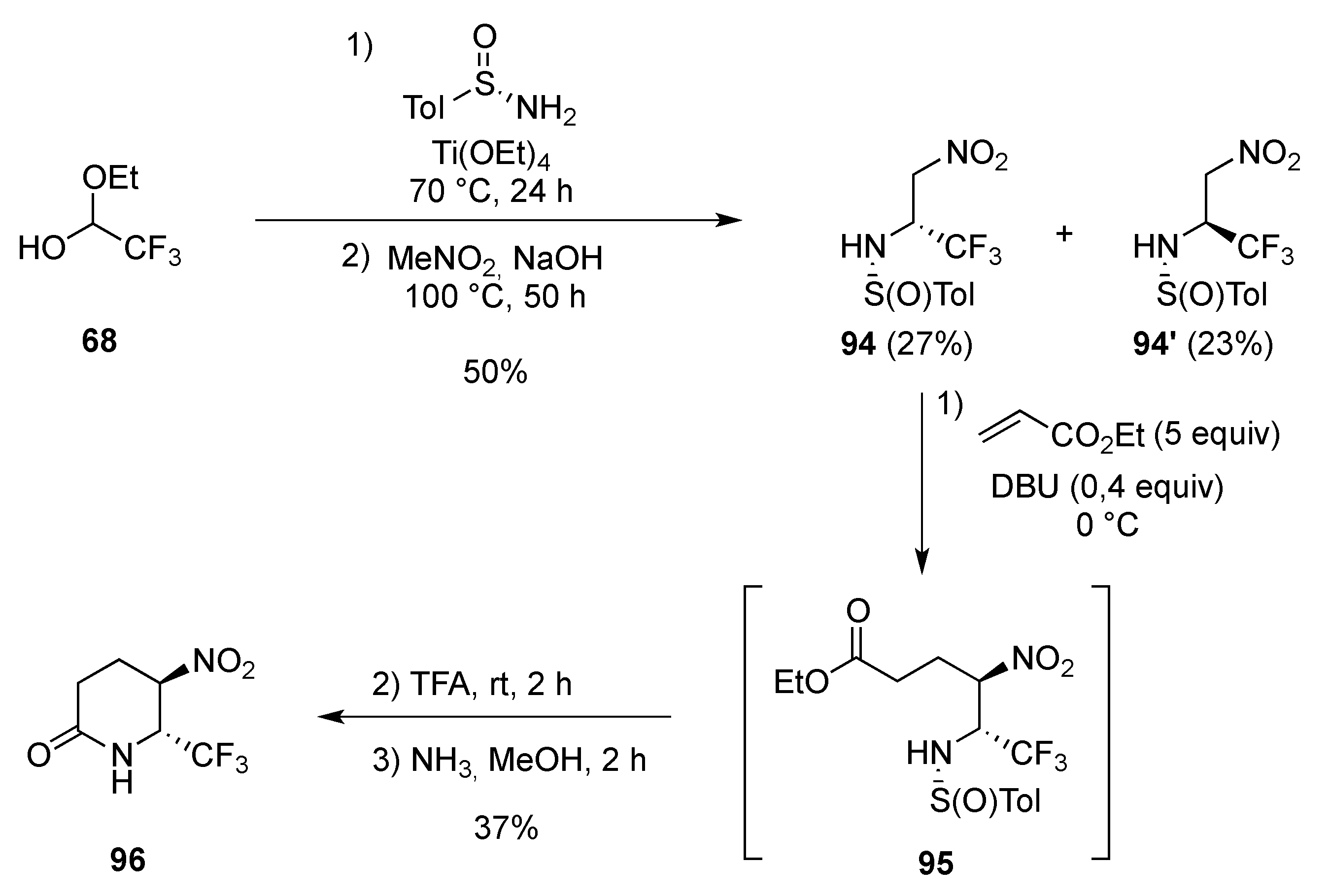
3.1.4. Electrophilic-Induced Cyclization
3.1.5. Nucleophilic Attack of Iminium Ions
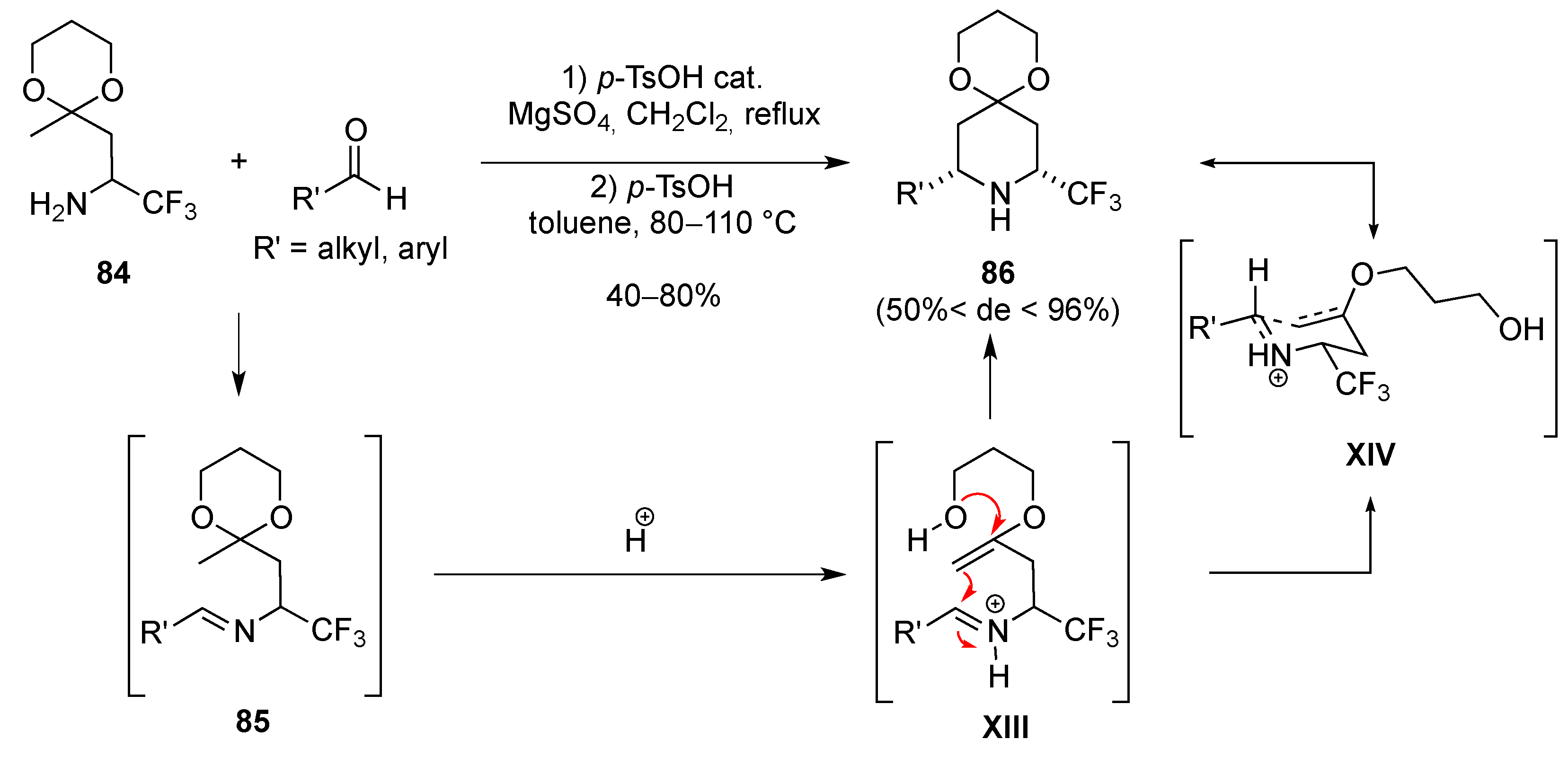
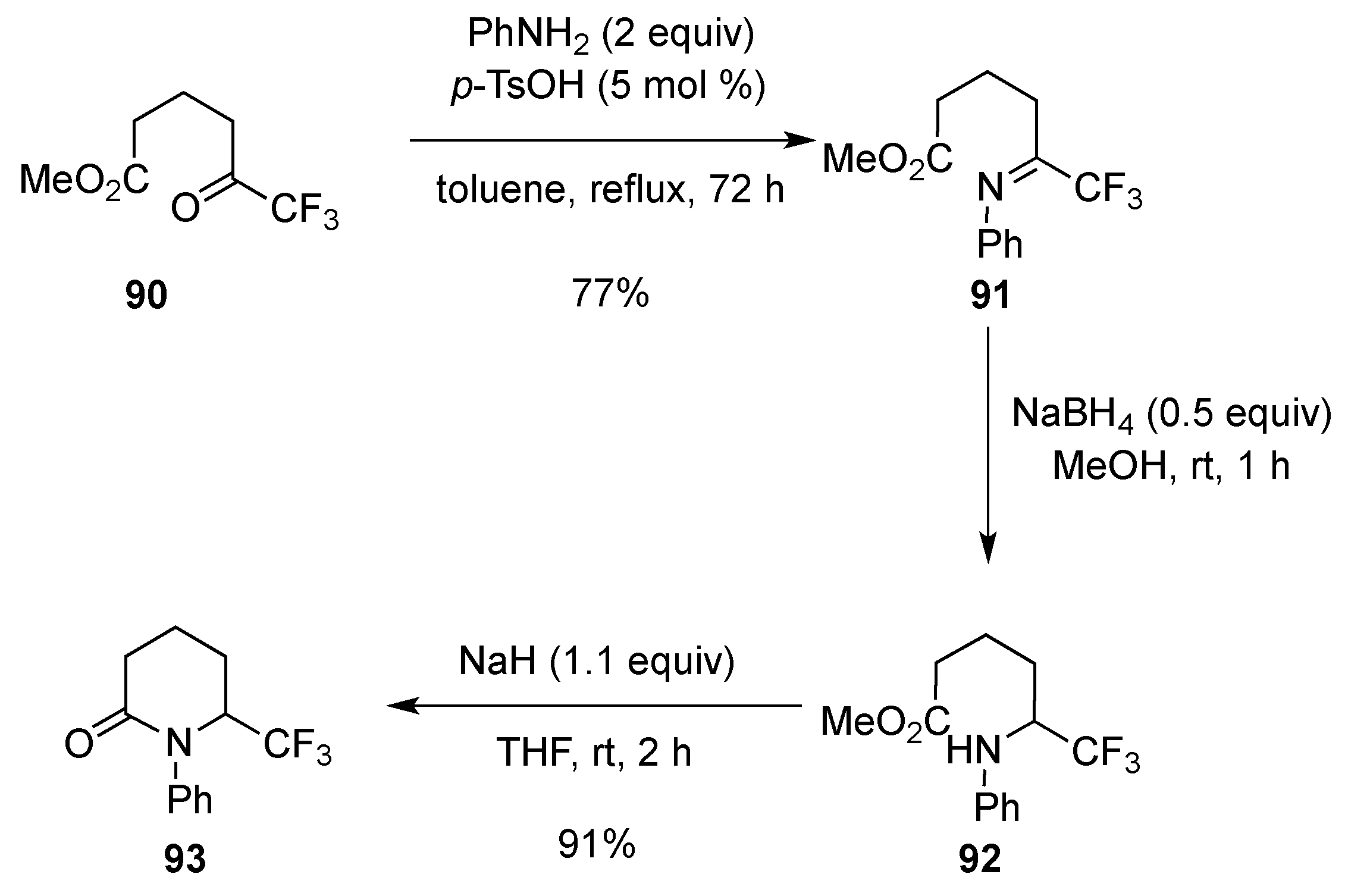
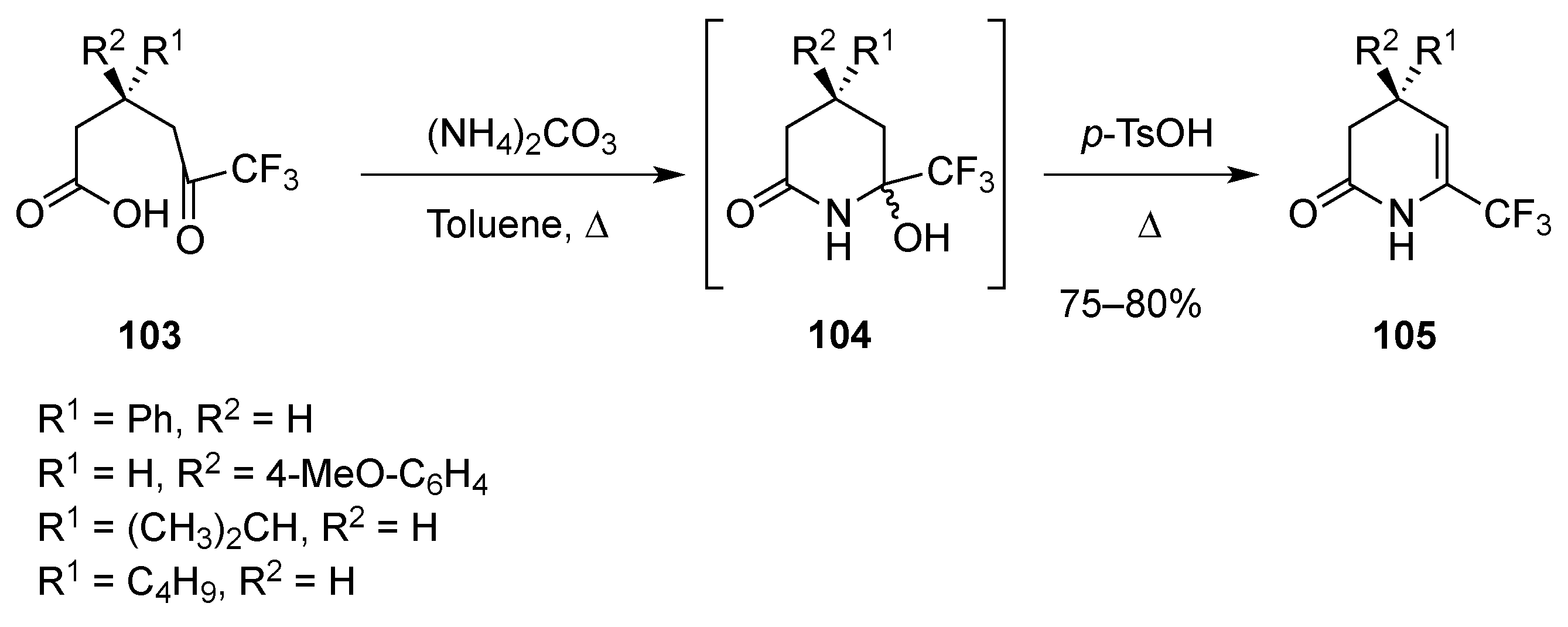
3.1.6. Intramolecular Condensation of Aminoketones and Aminoesters
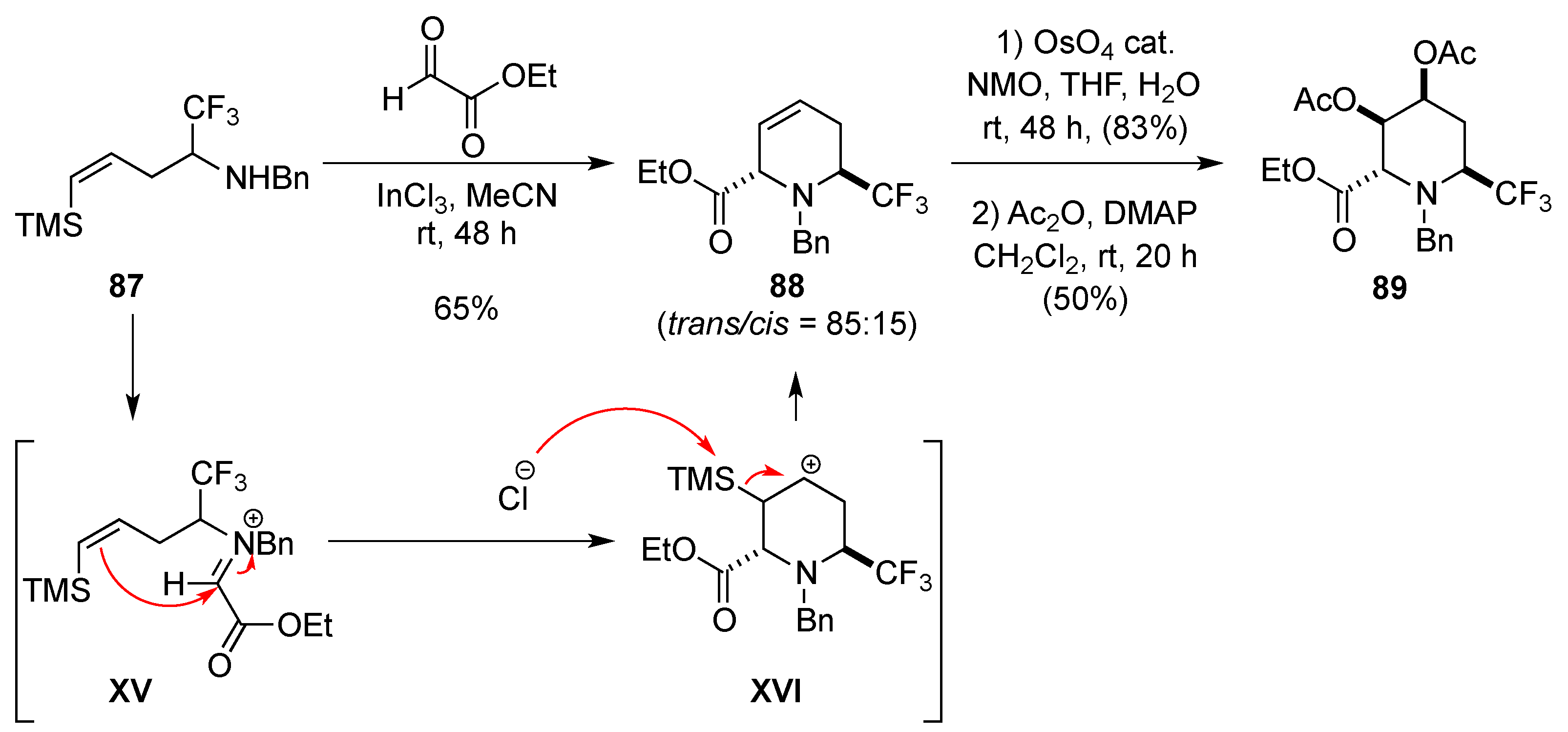
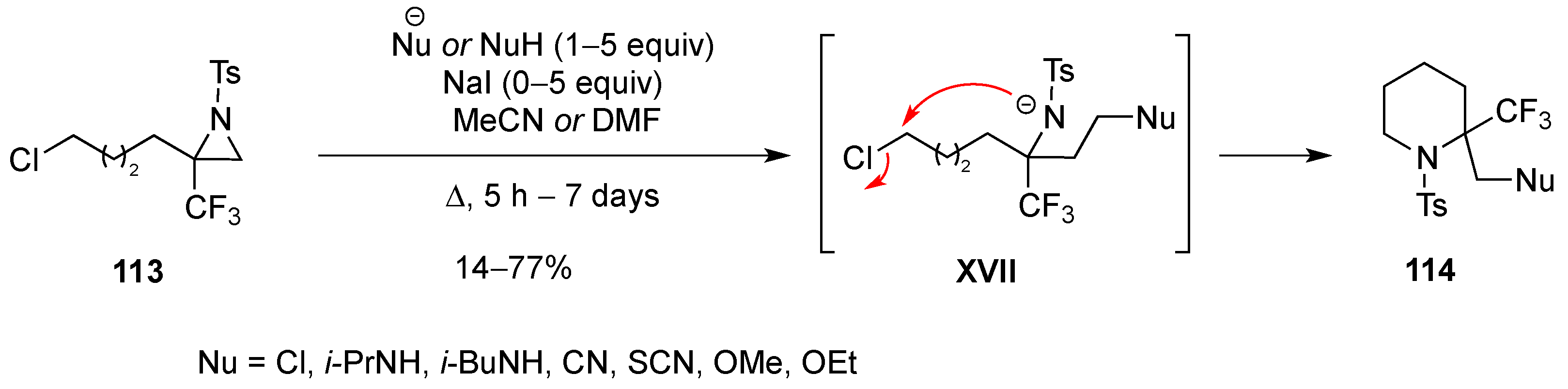
3.1.7. Intramolecular Nucleophilic Substitution
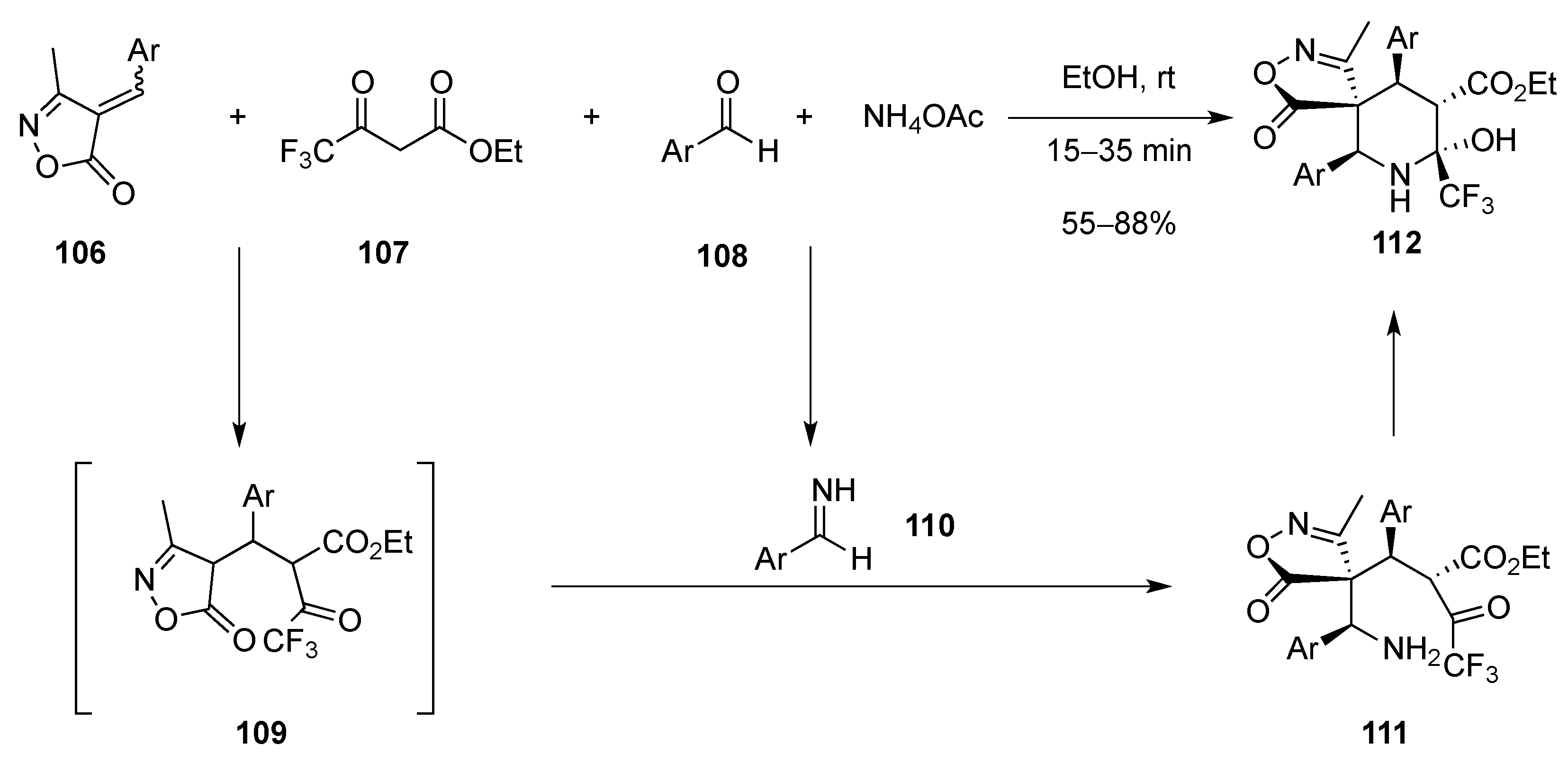
3.1.8. Intramolecular Nucleophilic Attack of Aldehydes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Laschat, S.; Dickner, T. Stereoselective synthesis of piperidines. Synthesis 2000, 1781–1813. [Google Scholar] [CrossRef]
- Toyooka, N.; Nemoto, H. Application of chiral building blocks to the synthesis of drugs. Drugs Fut. 2002, 27, 143–158. [Google Scholar] [CrossRef]
- Felpin, F.-X.; Lebreton, J. Recent advances in the total synthesis of piperidine and pyrrolidine natural alkaloids with ring-closing metathesis as a key step. Eur. J. Org. Chem. 2003, 3693–3712. [Google Scholar] [CrossRef]
- Basra, S.; Blechert, S. Ring rearrangement metathesis—A new concept in piperidine and pyrrolidine synthesis. Strategies Tactics Org. Synt. 2004, 4, 315–346. [Google Scholar]
- Pearson, M.S.M.; Mathe-Allainmat, M.; Fargeas, V.; Lebreton, J. Recent advances in the total synthesis of piperidine aza-sugars. Eur. J. Org. Chem. 2005, 2159–2191. [Google Scholar] [CrossRef]
- Vicario, J.L.; Badia, D.; Carrillo, L.; Ruiz, N.; Reyes, E. New methods for the asymmetric synthesis of piperidines and pyrrolidines: Chiral auxiliaries and asymmetric organocatalysis. Targets Heterocycl. Syst. 2008, 12, 302–327. [Google Scholar] [CrossRef]
- Amat, M.; Perez, M.; Bosch, J. Stereoselective conjugate addition reactions to phenylglycinol-derived, unsaturated oxazolopiperidone lactams. Chem. Eur. J. 2011, 17, 7724–7732. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F. Trifluoromethyl nitrogen heterocycles: Synthetic aspects and potential biological targets. Chem. Commun. 2016, 52, 3077–3094. [Google Scholar] [CrossRef] [PubMed]
- Raasch, M.S. Chemistry of sulfur tetrafluoride. IX. Reaction with amino acids in hydrogen fluoride. J. Org. Chem. 1962, 27, 1406–1409. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.C.; Harmange, J.-C.; Vaswani, R.G. Preparation of Substituted Indolecarboxamides as Modulators of Methyl Modifying Enzymes. WO 2016130396, 18 August 2016. [Google Scholar]
- Rice, K. Preparation of Benzoxazepines as PI3K/mTOR Inhibitors Useful in the Treatment of Cancer. WO 2012071509, 31 May 2012. [Google Scholar]
- Rice, K.; Markby, D. Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use as Antitumor Agents and Manufacture. WO 2012068106, 24 May 2012. [Google Scholar]
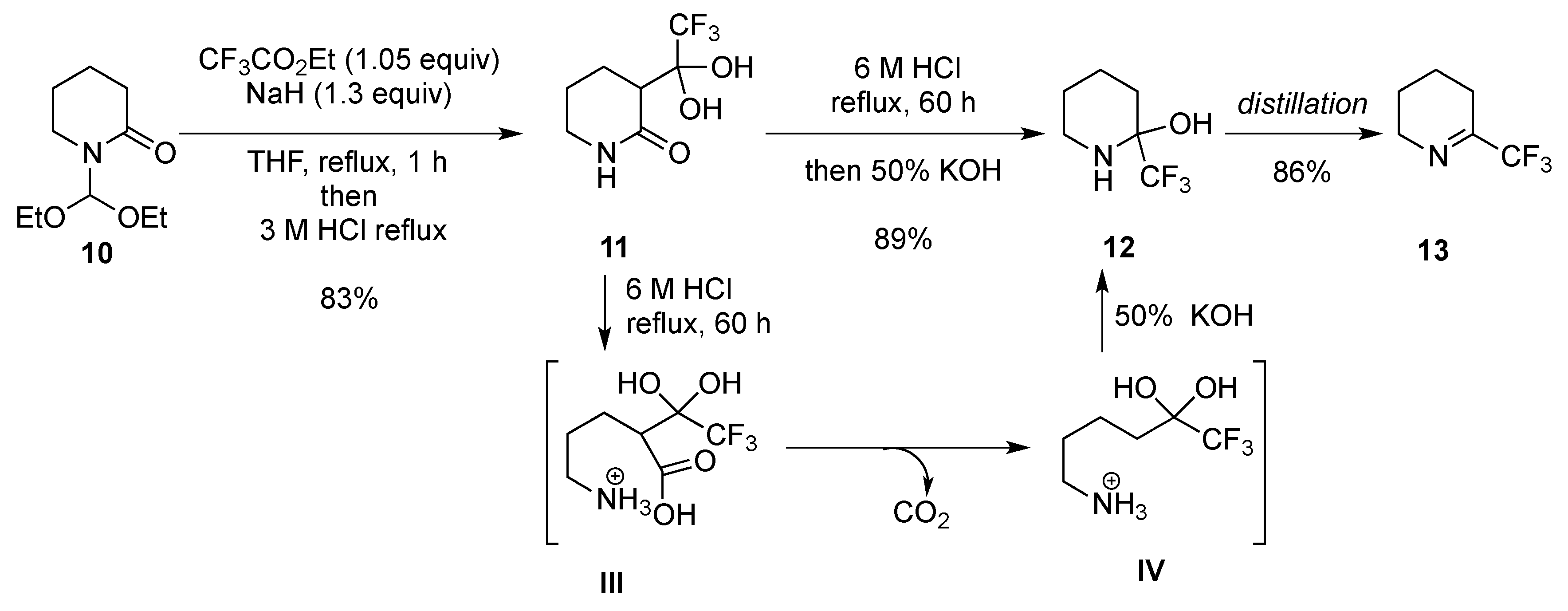
- Tolmachova, N.A.; Dolovanyuk, V.G.; Gerus, I.I.; Kondratov, I.S.; Polovinko, V.V.; Bergander, K.; Haufe, G. Catalytic hydrogenation of 3-amino-6-(trifluoromethyl)-5,6-dihydropyridin-2(1H)-ones and its use in the synthesis of trifluoromethyl-containing mimetics of ornithine and thalidomide. Synthesis 2011, 1149–1156. [Google Scholar] [CrossRef]
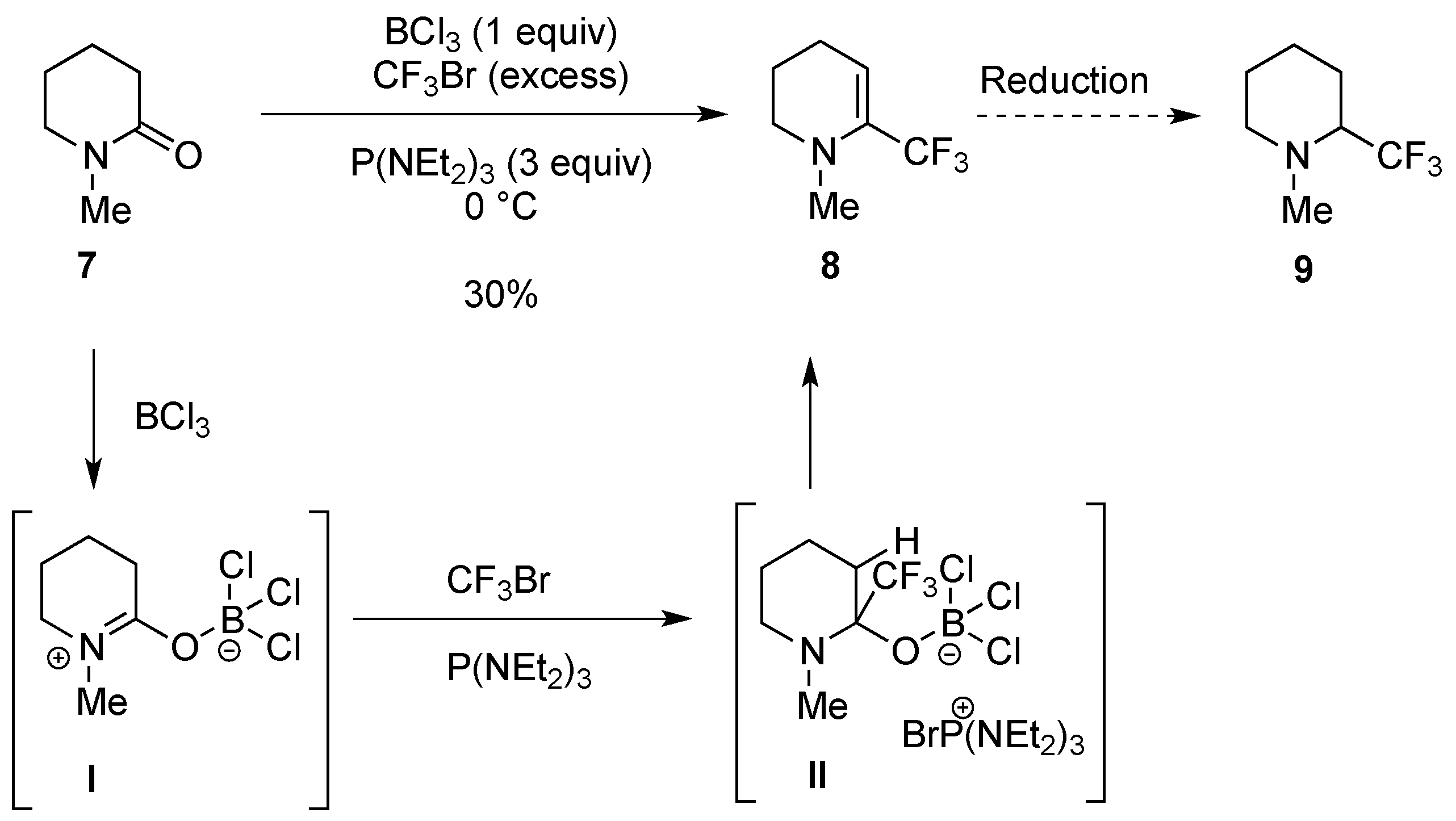
- Bürger, H.; Dittmar, T.; Pawelke, G. A simple one-pot synthesis of α-trifluoromethyl-substituted enamines by C-trifluoromethylation of dialkylamides with P(NEt2)3/CF3Br. J. Fluor. Chem. 1995, 70, 89–93. [Google Scholar] [CrossRef]
- Shevchenko, N.E.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Practical synthesis of α-perfluoroalkyl cyclic imines and amines. Synthesis 2010, 120–126. [Google Scholar] [CrossRef]
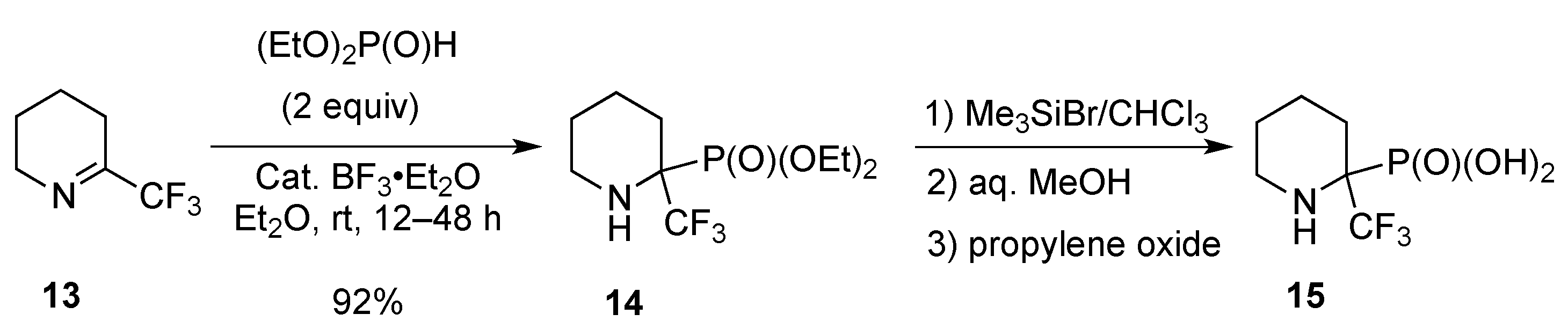
- Odinets, I.L.; Artyushin, O.I.; Lyssenko, K.A.; Shevchenko, N.E.; Nenajdenko, V.G.; Röschenthaler, G.-V. Facile synthesis of cyclic α-perfluoroalkyl-α-aminophosphonates. J. Fluor. Chem. 2009, 130, 662–666. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Shevchenko, N.E.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Efficient multicomponent synthesis of α-trifluoromethyl proline, homoproline, and azepan carboxylic acid dipeptides. Synlett 2009, 403–406. [Google Scholar] [CrossRef]
- Shmatova, O.I.; Nenajdenko, V.G. Tetrazole-substituted five-, six-, and seven-membered cyclic amines bearing perfluoroalkyl groups—efficient synthesis by azido-Ugi reaction. Eur. J. Org. Chem. 2013, 6397–6403. [Google Scholar] [CrossRef]
- Shmatova, O.I.; Shevchenko, N.E.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Highly β-regioselective Friedel-Crafts aminoalkylation of pyrroles with cyclic perfluoroalkylated imines. Eur. J. Org. Chem. 2013, 3049–3058. [Google Scholar] [CrossRef]
- Shmatova, O.I.; Shevchenko, N.E.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Friedel-Crafts alkylation of natural amino acid-derived pyrroles with CF3-substituted cyclic imines. Mendeleev Commun. 2013, 23, 92–93. [Google Scholar] [CrossRef]
- Shevchenko, N.E.; Shmatova, O.I.; Balenkova, E.S.; Röschenthaler, G.-V.; Nenajdenko, V.G. Aminoalkylation of indoles with α-polyfluoroalkylated cyclic imines. Eur. J. Org. Chem. 2013, 2237–2245. [Google Scholar] [CrossRef]
- Shevchenko, N.E.; Vlasov, K.; Nenajdenko, V.G.; Röschenthaler, G.-V. The reaction of cyclic imines with the Ruppert-Prakash reagent. Facile approach to α-trifluoromethylated nornicotine, anabasine, and homoanabasine. Tetrahedron 2011, 67, 69–74. [Google Scholar] [CrossRef]
- Cossy, J.; Gomez Pardo, D. Enantioselective ring expansion via aziridinium intermediates. Synthesis of substituted piperidines from substituted pyrrolidines. Synthetic applications. Targets Heterocycl. Syst. 2002, 6, 1–26. [Google Scholar] [CrossRef]
- Cossy, J.; Gomez Pardo, D.; Dumas, C.; Mirguet, O.; Dechamps, I.; Métro, T.-X.; Burger, B.; Roudeau, R.; Appenzeller, J.; Cochi, A. Rearrangement of β-amino alcohols and application to the synthesis of biologically active compounds. Chirality 2009, 21, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Tynoshenko, D.D. Ring expansions of 1-azabicyclo[n.1.0]alkanes. Recent developments. Arkivoc 2011, 329–345. [Google Scholar] [CrossRef]
- Cochi, A.; Gomez Pardo, D.; Cossy, J. Access to optically active 3-aminopiperidines by ring expansion of prolinols: Thermodynamic versus kinetic control. Eur. J. Org. Chem. 2012, 2023–2040. [Google Scholar] [CrossRef]
- Gomez Pardo, D.; Cossy, J. Access to optically active 3-substituted piperidines by ring expansion of prolinols and derivatives. Chem. Eur. J. 2014, 20, 4516–4525. [Google Scholar] [CrossRef] [PubMed]
- Dolfen, J.; Yadav, N.N.; De Kimpe, N.; D'hooghe, M.; Ha, H.-J. Bicyclic aziridinium ions in azaheterocyclic chemistry—Preparation and synthetic application of 1-azoniabicyclo[n.1.0]alkanes. Adv. Synth. Catal. 2016, 358, 3485–3511. [Google Scholar] [CrossRef]
- Rioton, S.; Orliac, A.; Antoun, Z.; Bidault, R.; Gomez Pardo, D.; Cossy, J. Stereoselective rearrangement of (trifluoromethyl)prolinols to enantioenriched 3-substituted 2-(trifluoromethyl)piperidines. Org. Lett. 2015, 17, 2916–2919. [Google Scholar] [CrossRef] [PubMed]
- Orliac, A. Synthèse et propriétés physico‑chimiques des fluoro-pipéridines. XtalFluor-E et synthèse d′amides. Ph.D. Thesis, Université Pierre et Marie Curie, Paris, France, 13 November 2013. [Google Scholar]
- Schmalz, H.-G. Catalytic ring-closing metathesis: a new, powerful technique for carbon-carbon coupling in organic synthesis. Angew. Chem. Int. Ed. 1995, 34, 1833–1836. [Google Scholar] [CrossRef]
- Grubbs, R.H.; Miller, S.J.; Fu, G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995, 28, 446–452. [Google Scholar] [CrossRef]
- Schuster, M.; Blechert, S. Olefin metathesis in organic chemistry. Angew. Chem. Int. Ed. 1997, 36, 2037–2056. [Google Scholar] [CrossRef]
- Armstrong, S.K. Ring-closing diene metathesis in organic synthesis. J. Chem. Soc., Perkin Trans. 1: Org. Bio-Org. Chem. 1998, 371–388. [Google Scholar] [CrossRef]
- Wright, D.L. Application of olefin metathesis to organic synthesis. Curr. Org. Chem. 1999, 3, 211–240. [Google Scholar]
- Billard, T.; Langlois, B.R. Reactivity of stable trifluoroacetaldehyde hemiaminals. 2. Generation and synthetic potentialities of fluorinated iminiums. J. Org. Chem. 2002, 67, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Gille, S.; Ferry, A.; Billard, T.; Langlois, B.R. Synthesis of α-trifluoromethylated nitrogen heterocycles. J. Org. Chem. 2003, 68, 8932–8935. [Google Scholar] [CrossRef] [PubMed]
- Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. A one-pot synthesis of doubly unsaturated trifluoromethyl amines: Easy access to CF3-substituted piperidines. Eur. J. Org. Chem. 2005, 1258–1265. [Google Scholar] [CrossRef]
- Fürstner, A.; Picquet, M.; Bruneau, C.; Dixneuf, P.H. Cationic ruthenium allenylidene complexes as a new class of performing catalysts for ring-closing metathesis. Chem. Commun. 1998, 1315–1316. [Google Scholar] [CrossRef]
- Osipov, S.N.; Bruneau, C.; Picquet, M.; Kolomiets, A.F.; Dixneuf, P.H. Synthesis of fluorine-containing cyclic amino acid derivatives via ring-closing olefin metathesis. Chem. Commun. 1998, 2053–2054. [Google Scholar] [CrossRef]
- Osipov, S.N.; Artyushin, O.I.; Kolomiets, A.F.; Bruneau, C.; Dixneuf, P.H. α-CF3-substituted phosphorus containing analogs of α-amino acids. Novel six- and seven-membered α-amino phosphonates via ring-closing metathesis with LnRu=C=C=CR2 precatalyst. Synlett 2000, 1031–1033. [Google Scholar] [CrossRef]
- Osipov, S.N.; Artyushin, O.I.; Kolomiets, A.F.; Bruneau, C.; Picquet, M.; Dixneuf, P.H. Synthesis of fluorine-containing cyclic α-amino acid and α-amino phosphonate derivatives by alkene metathesis. Eur. J. Org. Chem. 2001, 3891–3897. [Google Scholar] [CrossRef]
- Vorobyeva, D.V.; Mailyan, A.K.; Peregudov, A.S.; Karimova, N.M.; Vasilyeva, T.P.; Bushmarinov, I.S.; Bruneau, C.; Dixneuf, P.H.; Osipov, S.N. Synthesis of functionalized CF3-containing heterocycles via [2,3]-sigmatropic rearrangement and sequential catalytic carbocyclization. Tetrahedron 2011, 67, 3524–3532. [Google Scholar] [CrossRef]
- Osipov, S.N.; Kobelikova, N.M.; Shchetnikov, G.T.; Kolomiets, A.F.; Bruneau, C.; Dixneuf, P.H. Novel synthesis of cyclic α-amino acid esters via ene reaction and ruthenium-catalyzed ring rearrangement. Synlett 2001, 621–622. [Google Scholar] [CrossRef]
- Mailyan, A.K.; Krylov, I.M.; Bruneau, C.; Dixneuf, P.H.; Osipov, S.N. Access to cyclic α-CF3-substituted α-amino acid derivatives by ring-closing metathesis of functionalized 1,7-enynes. Eur. J. Org. Chem. 2013, 5353–5363. [Google Scholar] [CrossRef]
- Mailyan, A.K.; Krylov, I.M.; Bruneau, C.; Dixneuf, P.H.; Osipov, S.N. Thermal [2 + 2]-cycloaddition of CF3-substituted allenynes: Access to novel cyclobutene-containing α-amino acids. Synlett 2011, 2321–2324. [Google Scholar] [CrossRef]
- Ferry, A.; Billard, T.; Langlois, B.R. Synthesis of α-trifluoromethylated nitrogen bicycles. Synlett 2005, 1027–1029. [Google Scholar] [CrossRef]
- Kobel′kova, N.M.; Osipov, S.N.; Kolomiets, A.F. Aza Diels-Alder reactions of methyl trifluoropyruvate sulfonyl- and phosphorylimines with 1,3-dienes. Russ. Chem. Bull. Int. Ed. 2002, 51, 1298–1302. [Google Scholar] [CrossRef]
- Li, P.; Liu, L.-J.; Liu, J.-T. Per(poly)fluoroalkanesulfinamides assisted diastereoselective three-component inverse-electron-demand aza Diels-Alder reaction. Org. Biomol. Chem. 2011, 9, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Monteagudo, S.; Sanchez-Rosello, M.; Flores, S.; Barrio, P.; Del Pozo, C. N-Sulfinyl amines as a nitrogen source in the asymmetric intramolecular aza-Michael reaction: Total synthesis of (−)-pinidinol. Chem. Eur. J. 2010, 16, 9835–9845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cha, L.; Li, L.; Hu, Y.; Li, Y.; Zha, Z.; Wang, Z. Asymmetric formal aza-Diels-Alder reaction of trifluoromethyl hemiaminals with enones catalyzed by primary amines. J. Org. Chem. 2016, 81, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Albert, L.; Aceña, J.L.; Sanz-Cervera, J.F.; Asensio, A. An Efficient entry to optically active anti- and syn-β-amino-α-trifluoromethyl alcohols. Org. Lett. 2008, 10, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Albert, L.; Mateu, N.; Chiva, G.; Miro, J.; Gonzalez, J.; Aceña, J.L. Stereoselective access to fluorinated and non-fluorinated quaternary piperidines: Synthesis of pipecolic acid and imino-sugar derivatives. Chem. Eur. J. 2012, 18, 3753–3764. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xu, B.; Hammond, G.B. Synthesis of α-CN and α-CF3 N-heterocycles through tandem nucleophilic additions. Org. Lett. 2011, 13, 3450–3453. [Google Scholar] [CrossRef] [PubMed]
- Bariau, A.; Roblin, J.-P.; Troin, Y.; Canet, J.-L. A simple stereoselective access to α-trifluoromethylated piperidines. Synlett 2005, 1731–1733. [Google Scholar] [CrossRef]
- Bariau, A.; Jatoi, W.B.; Calinaud, P.; Troin, Y.; Canet, J.-L. A simple stereoselective route to α-trifluoromethyl analogues of piperidine alkaloids. Eur. J. Org. Chem. 2006, 3421–3433. [Google Scholar] [CrossRef]
- Ciblat, S.; Besse, P.; Canet, J.-L.; Troin, Y.; Veschambre, H.; Gelas, J. A practical asymmetric synthesis of 2,6-cis-disubstituted piperidines. Tetrahedron: Asymmetry 1999, 10, 2225–2235. [Google Scholar] [CrossRef]
- Dobbs, A.P.; Parker, R.J.; Skidmore, J. Rapid access to CF3-containing heterocycles. Tetrahedron Lett. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Wan, W.; Hou, J.; Jiang, H.; Yuan, Z.; Ma, G.; Zhao, G.; Hao, J. Reductive amination/cyclization of ω-trifluoromethyl keto esters to trifluoromethylated δ-amino alcohols and lactams. Eur. J. Org. Chem. 2010, 1778–1786. [Google Scholar] [CrossRef]
- García Ruano, J.L.; De Haro, T.; Singh, R.; Belén Cid, M. An efficient method for the synthesis of nitropiperidones. J. Org. Chem. 2008, 73, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Spanedda, M.V.; Ourévitch, M.; Crousse, B.; Bégué, J.-P.; Bonnet-Delpon, D. Vinylogous Mannich reactions. additions of trimethylsilyloxyfuran to fluorinated aldimines. Tetrahedron Lett. 2004, 45, 5023–5025. [Google Scholar] [CrossRef]
- Okano, T.; Fumoto, M.; Kusukawa, T.; Fujita, M. Synthesis of optically active trifluoromethylated indolizidine derivatives via stereoselective radical cyclization. Org. Lett. 2002, 4, 1571–1573. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wang, Y.; Zhu, Y.; Zhang, M.; Song, L.; Deng, H. A one-pot, multicomponent synthesis of trifluoromethylated spiropiperidines under catalyst-free conditions. Synthesis 2016, 48, 3527–3536. [Google Scholar]
- Dolfen, J.; Kenis, S.; Van Hecke, K.; De Kimpe, N.; D′hooghe, M. Selective synthesis of functionalized trifluoromethylated pyrrolidines, piperidines, and azepanes starting from 1-tosyl-2-(trifluoromethyl)aziridine. Chem. Eur. J. 2014, 20, 10650–10653. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.P.; West, T.H.; McGarrigle, E.M.; Aggarwal, V.K. Diastereoselective synthesis of CF3-substituted, epoxide-fused heterocycles with β-(trifluoromethyl)vinylsulfonium salts. Org. Lett. 2012, 14, 6370–6373. [Google Scholar] [CrossRef] [PubMed]






































| Entry | R | R1 | R2 | R3 | T (°C) | t (h) | Yield |
|---|---|---|---|---|---|---|---|
| 1 | SO2Ph | Me | Me | H | –30 to 20 | 13 | 71% |
| 2 | SO2Me | Me | Me | H | –30 to 20 | 14 | 91% |
| 3 | P(O)(OEt)2 | Me | Me | H | 20 | 120 | 95% |
| 4 | SO2Ph | H | Me | H | –30 to 20 | 13 | 81% |
| 5 | SO2Me | H | Me | H | –30 to 20 | 13 | 97% |
| 6 | P(O)(OEt)2 | H | Me | H | 20 | 150 | 68% |
| 7 | SO2Ph | H | H | Me | 4 | 120 | 75% |
| 8 | P(O)(OEt)2 | H | H | Me | 20 | 720 | 89% |

{kind=link}
{kind=link}
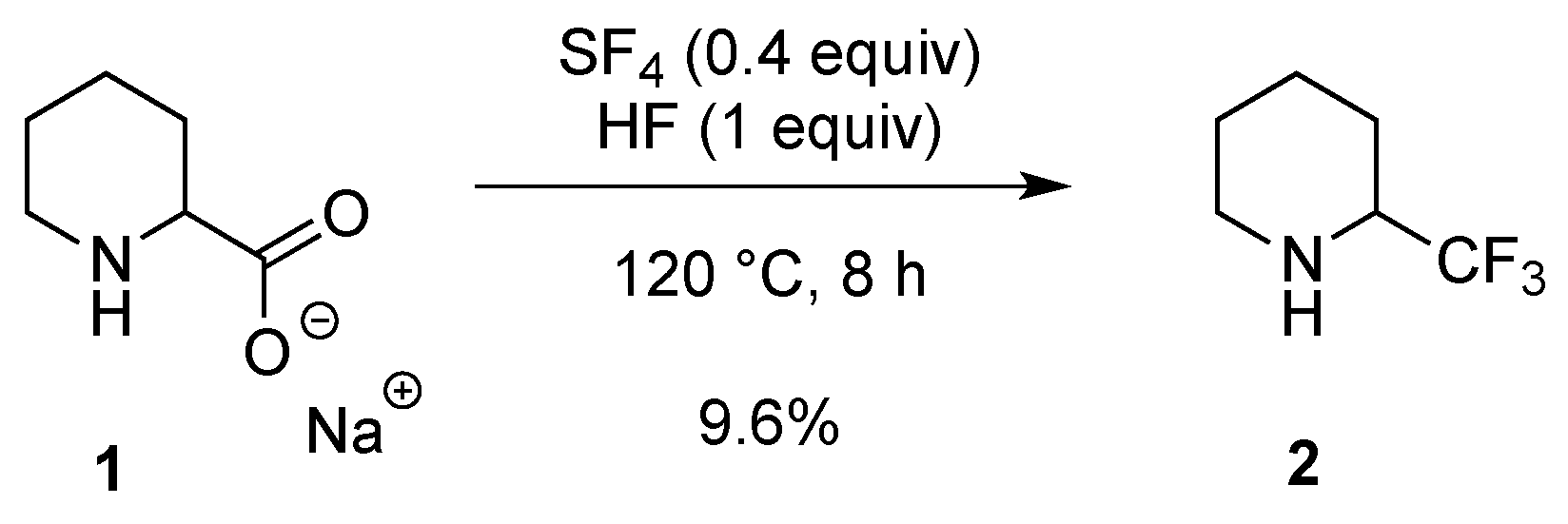{kind=link}
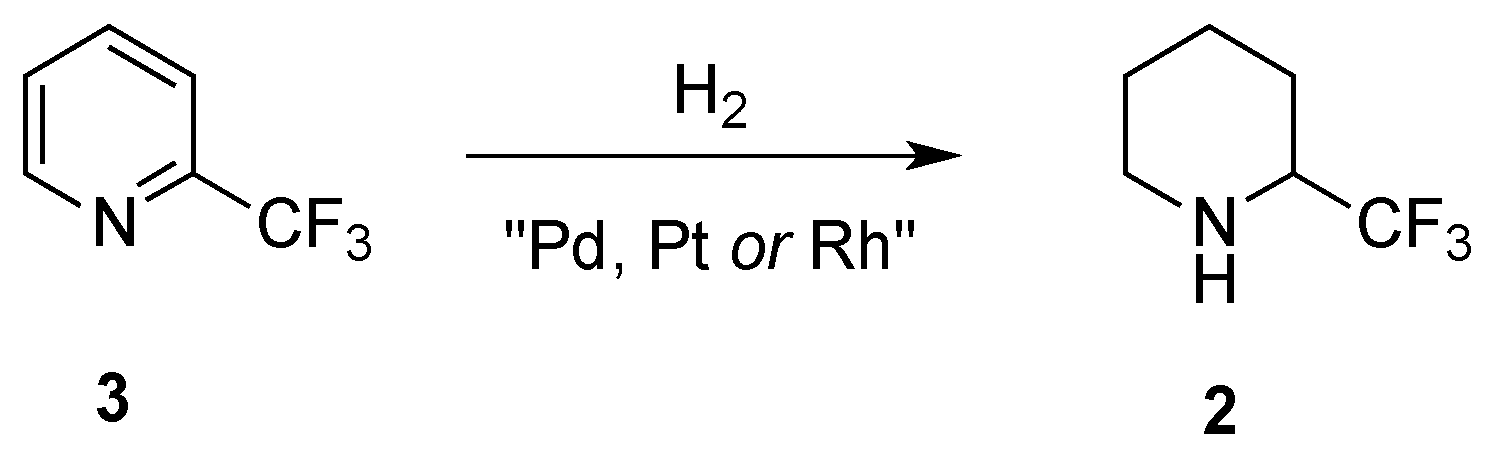{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
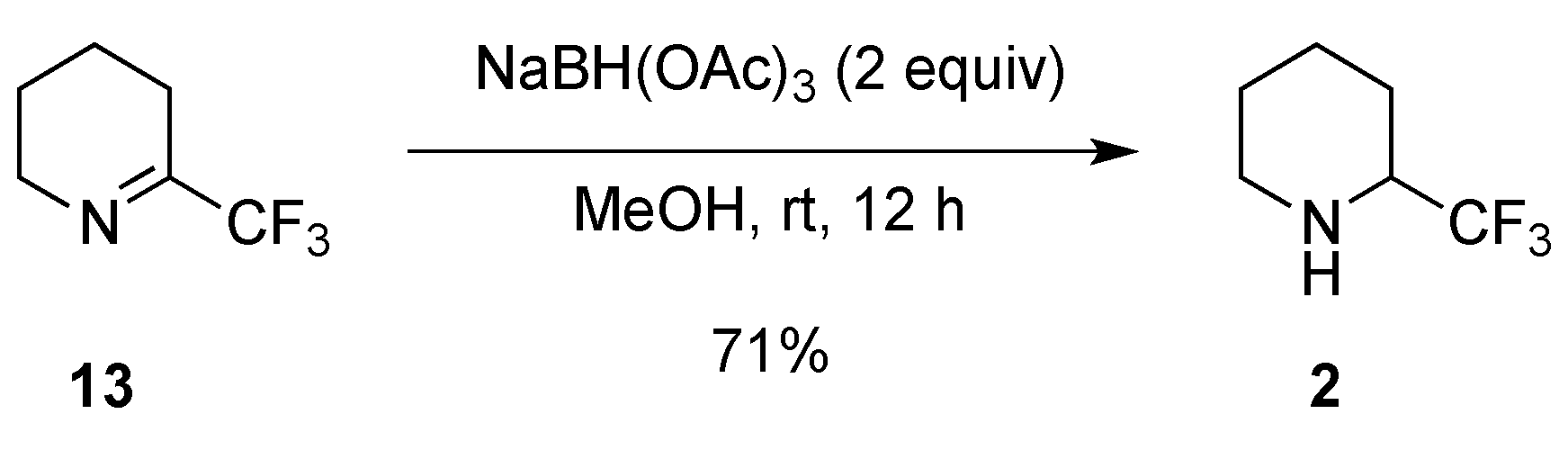{kind=link}
{kind=link}
{kind=link}
{kind=link}
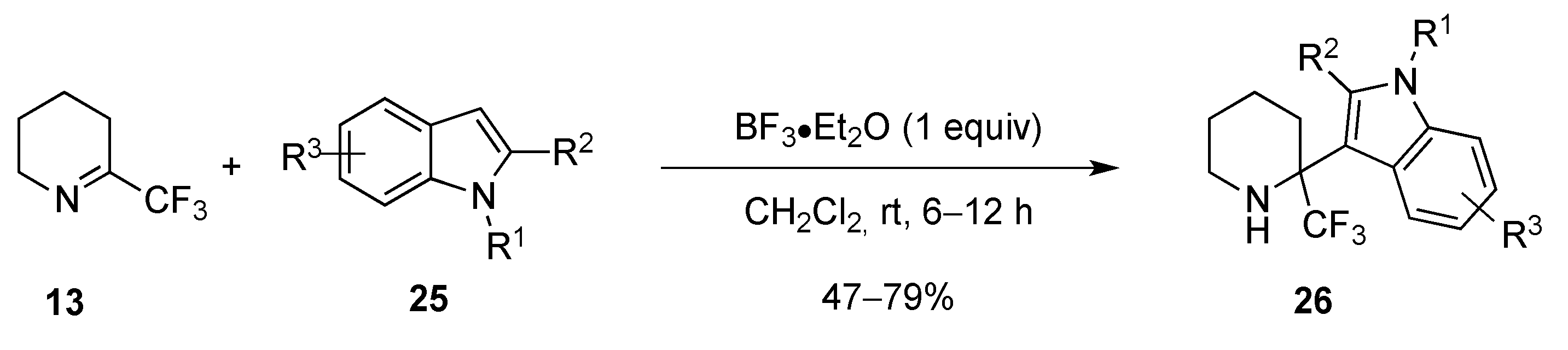{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rioton, S.; Pardo, D.G.; Cossy, J. Synthesis of Substituted α-Trifluoromethyl Piperidinic Derivatives. Molecules 2017, 22, 483. https://doi.org/10.3390/molecules22030483
Rioton S, Pardo DG, Cossy J. Synthesis of Substituted α-Trifluoromethyl Piperidinic Derivatives. Molecules. 2017; 22(3):483. https://doi.org/10.3390/molecules22030483
Chicago/Turabian StyleRioton, Sarah, Domingo Gomez Pardo, and Janine Cossy. 2017. "Synthesis of Substituted α-Trifluoromethyl Piperidinic Derivatives" Molecules 22, no. 3: 483. https://doi.org/10.3390/molecules22030483
APA StyleRioton, S., Pardo, D. G., & Cossy, J. (2017). Synthesis of Substituted α-Trifluoromethyl Piperidinic Derivatives. Molecules, 22(3), 483. https://doi.org/10.3390/molecules22030483