
A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives

Abstract
:
1. Introduction
2. Results
2.1. Synthesis of the Levoglucosan Derivatives
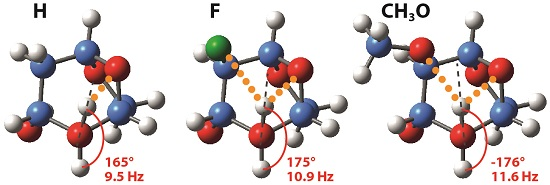
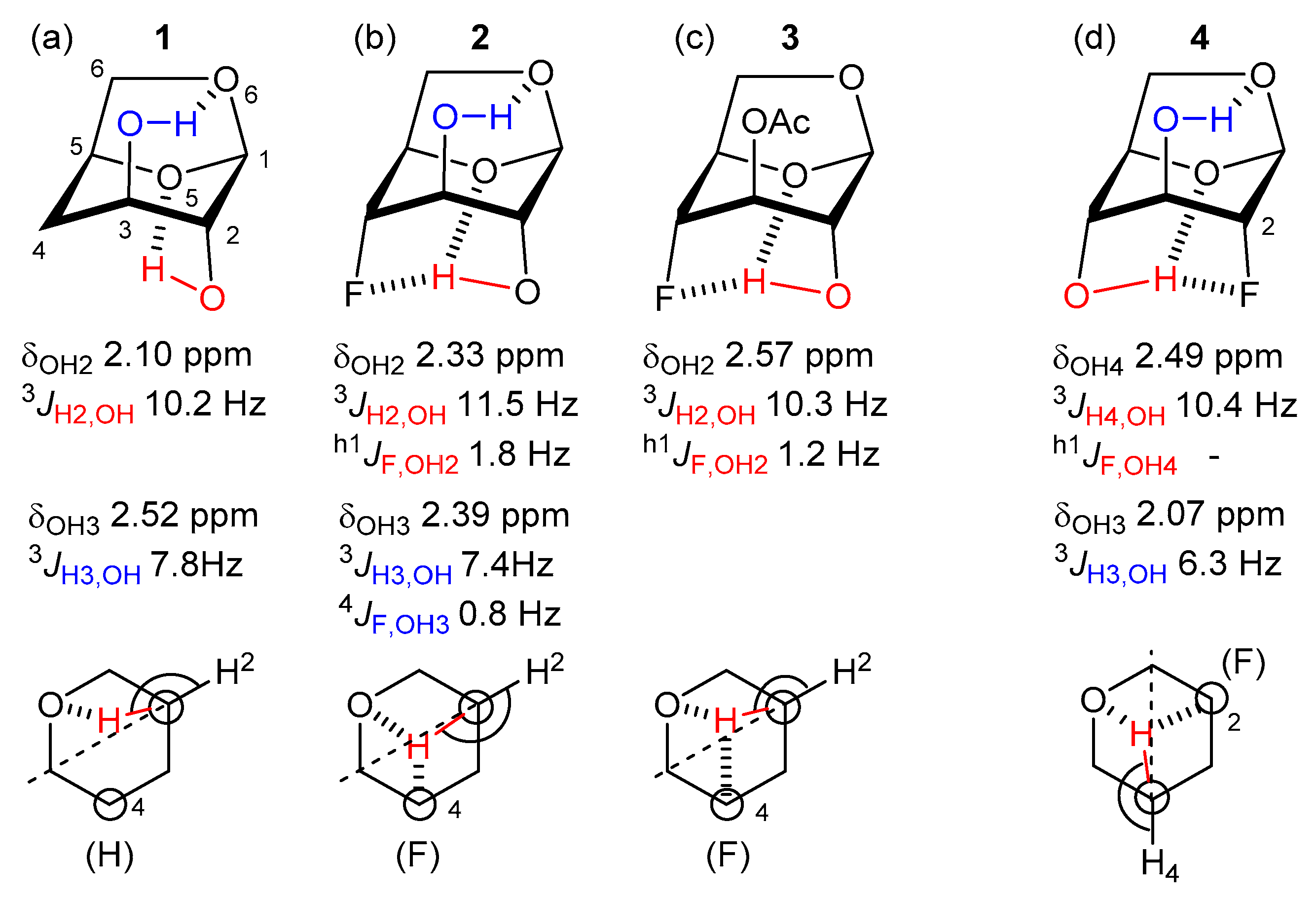
2.2. Experimental and Theoretical NMR Features
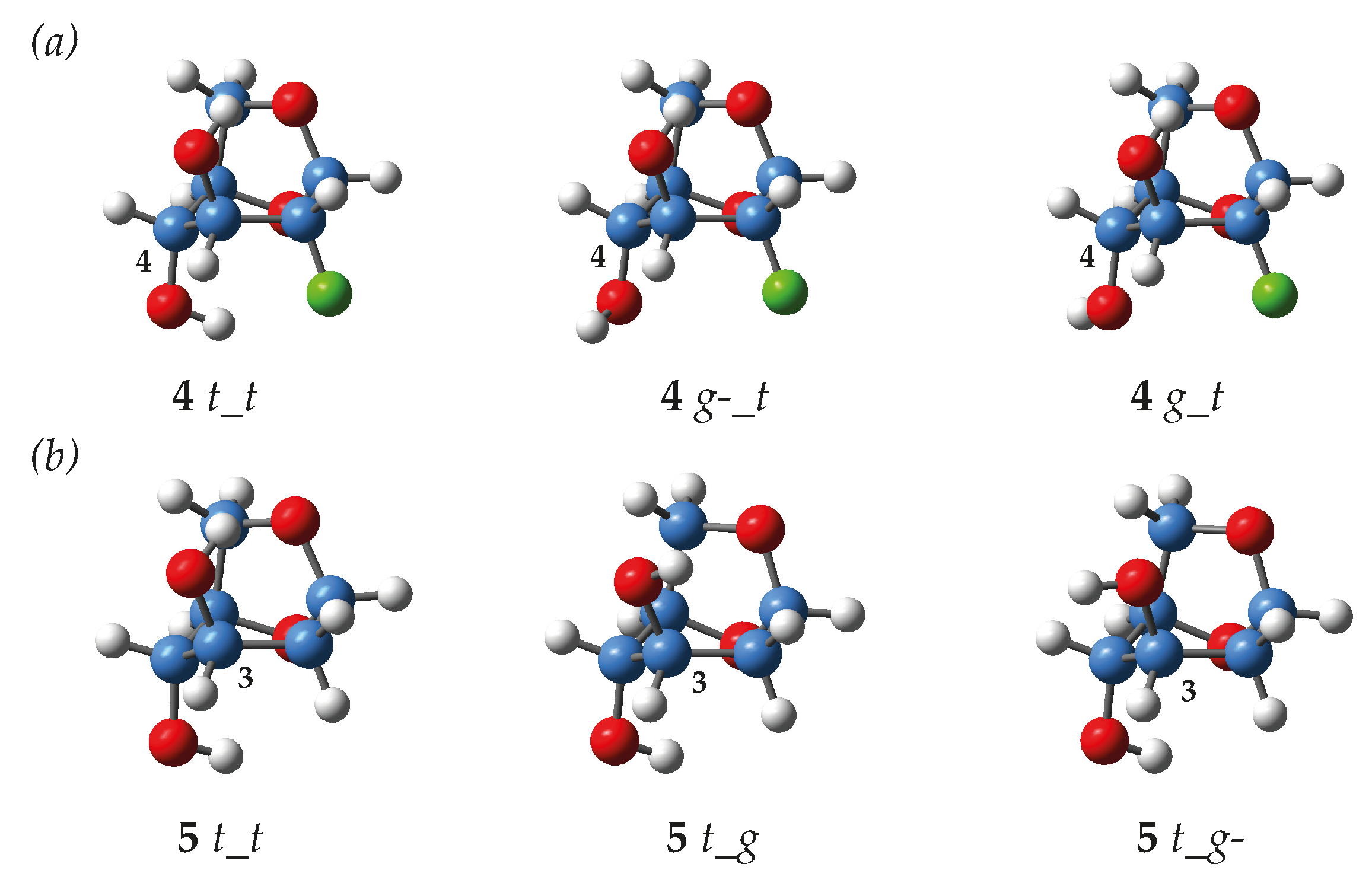
2.3. Conformational Analysis of the Levoglucosans
2.4. Levoglucosans Wavefunction Analyses
3. Discussion
3.1. Description of the Intramolecular OH4 Interactions
3.1.1. IMHB Involving OH4
3.1.2. Interplay between OH3 Features and IMHB Involving OH4
3.2. Description of the Intramolecular OH3 Interactions
4. Materials and Methods
4.1. NMR Data Acquisition
4.2. Quantum Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schneider, H.-J. Hydrogen bonds with fluorine. Studies in solution, in gas phase and by computations, conflicting conclusions from crystallographic analyses. Chem. Sci. 2012, 3, 1381–1394. [Google Scholar] [CrossRef]
- Howard, J.A.K.; Hoy, V.J.; O’Hagan, D.; Smith, G.T. How good is fluorine as a hydrogen bond acceptor? Tetrahedron 1996, 52, 12613–12622. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Taylor, R. Organic fluorine hardly ever accepts hydrogen bonds. Chem. Eur. J. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Mehta, G.; Sen, S. Probing fluorine interactions in a polyhydroxylated environment: Conservation of a C-F···H-C recognition motif in presence of O-H···O hydrogen bonds. Eur. J. Org. Chem. 2010, 2010, 3387–3394. [Google Scholar] [CrossRef]
- Linclau, B.; Golten, S.; Light, M.; Sebban, M.; Oulyadi, H. The conformation of tetrafluorinated methyl galactoside anomers: Crystallographic and NMR studies. Carbohydr. Res. 2011, 346, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, M.A.; Vasella, A. Glycosylidene carbenes part 26. The intramolecular F…HO hydrogen bond of 1,3-diaxial 3-fluorocyclohexanols. Helv. Chim. Acta 1998, 81, 695–717. [Google Scholar] [CrossRef]
- Champagne, P.A.; Desroches, J.; Paquin, J.-F. Organic fluorine as a hydrogen-bond acceptor: Recent examples and applications. Synthesis 2015, 47, 306–322. [Google Scholar]
- Linclau, B.; Peron, F.; Bogdan, E.; Wells, N.; Wang, Z.; Compain, G.; Fontenelle, C.Q.; Galland, N.; Le Questel, J.-Y.; Graton, J. Intramolecular OH···Fluorine hydrogen bonding in saturated, acyclic fluorohydrins: The γ-fluoropropanol motif. Chem. Eur. J. 2015, 21, 17808–17816. [Google Scholar] [CrossRef] [PubMed]
- Bernet, B.; Vasella, A. Hydrogen bonding of fluorinated saccharides in solution: F acting as H-bond acceptor in a bifurcated H-bond of 4-fluorinated levoglucosans. Helv. Chim. Acta 2007, 90, 1874–1888. [Google Scholar] [CrossRef]
- Giuffredi, G.T.; Gouverneur, V.; Bernet, B. Intramolecular OH···FC hydrogen bonding in fluorinated carbohydrates: CHF is a better hydrogen bond acceptor than CF2. Angew. Chem. Int. Ed. 2013, 52, 10524–10528. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Invernizzi, C.; Vulpetti, A. Fluorine as a hydrogen-bond acceptor: Experimental evidence and computational calculations. Chem. Eur. J. 2014, 20, 11058–11068. [Google Scholar] [CrossRef] [PubMed]
- Roennols, J.; Manner, S.; Siegbahn, A.; Ellervik, U.; Widmalm, G. Exploration of conformational flexibility and hydrogen bonding of xylosides in different solvents, as a model system for enzyme active site interactions. Org. Biomol. Chem. 2013, 11, 5465–5472. [Google Scholar] [CrossRef] [PubMed]
- Graton, J.; Wang, Z.; Brossard, A.-M.; Goncalves Monteiro, D.; Le Questel, J.-Y.; Linclau, B. An unexpected and significantly lower hydrogen-bond-donating capacity of fluorohydrins compared to nonfluorinated alcohols. Angew. Chem. Int. Ed. 2012, 51, 6176–6180. [Google Scholar] [CrossRef] [PubMed]
- Laurence, C.; Brameld, K.A.; Graton, J.; Le Questel, J.-Y.; Renault, E. The pKBHX database: Toward a better understanding of hydrogen-bond basicity for medicinal chemists. J. Med. Chem. 2009, 52, 4073–4086. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Seib, P.A. 1,6-anhydro-2-deoxy-β-d-hexopyranoses. J. Chem. Soc. C 1969, 19, 2552–2559. [Google Scholar] [CrossRef]
- Sarda, P.; Cabrera Escribano, F.; Alves, R.J.; Olesker, A.; Lukacs, G. Stereospecific access to 2,3,4-trideoxy-2,3,4-trifluoro-d-glucose and d-galactose derivatives. J. Carbohydr. Chem. 1989, 8, 115–123. [Google Scholar] [CrossRef]
- Mtashobya, L.; Quiquempoix, L.; Linclau, B. The synthesis of mono- and difluorinated 2,3-dideoxy-d-glucopyranoses. J. Fluorine Chem. 2015, 171, 92–96. [Google Scholar] [CrossRef]
- Wollwage, P.C.; Seib, P.A. Thermal degradation of 2-O-methylcellulose. Carbohydr. Res. 1969, 10, 589–594. [Google Scholar] [CrossRef]
- Hori, H.; Nishida, Y.; Ohrui, H.; Meguro, H. Regioselective de-O-benzylation with lewis acids. J. Org. Chem. 1989, 54, 1346–1353. [Google Scholar] [CrossRef]
- Pacak, J.; Podesva, J.; Tocik, Z.; Cerny, M. Syntheses with anhydro sugars. XI. Preparation of 2-deoxy-fluoro-d-glucose and 2,4-dideoxy-2,4-difluoro-d-glucose. Collect. Czech. Chem. Commun. 1972, 37, 2589–2599. [Google Scholar] [CrossRef]
- Barford, A.D.; Foster, A.B.; Westwood, J.H.; Hall, L.D.; Johnson, R.N. Fluorinated carbohydrates. Carbohydr. Res. 1971, 19, 49–61. [Google Scholar] [CrossRef]
- Cormanich, R.A.; Freitas, M.P.; Tormena, C.F.; Rittner, R. The F···HO intramolecular hydrogen bond forming five-membered rings hardly appear in monocyclic organofluorine compounds. RSC Adv. 2012, 2, 4169–4174. [Google Scholar] [CrossRef]
- Cormanich, R.A.; Rittner, R.; Freitas, M.P.; Buhl, M. The seeming lack of CF···HO intramolecular hydrogen bonds in linear aliphatic fluoroalcohols in solution. Phys. Chem. Chem. Phys. 2014, 16, 19212–19217. [Google Scholar] [CrossRef] [PubMed]
- Graton, J.; Compain, G.; Besseau, F.; Bogdan, E.; Watts, J.M.; Mtashobya, L.; Wang, Z.; Weymouth-Wilson, A.; Galland, N.; Le Questel, J.-Y.; et al. Influence of alcohol β-fluorination on hydrogen-bond acidity of conformationally flexible substrates. Chem. Eur. J. 2017, 23, 2811–2819. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wilson, P.J.; Bradley, T.J.; Tozer, D.J. Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials. J. Chem. Phys. 2001, 115, 9233–9242. [Google Scholar] [CrossRef]
- Jensen, F. The basis set convergence of spin-spin coupling constants calculated by density functional methods. J. Chem. Theory Comput. 2006, 2, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Biegler-Koenig, F.W.; Schonbohm, J.; Bayles, D. AIM2000—A program to analyze and visualize atoms in molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Espinosa, E.; Lecomte, C.; Molins, E. Experimental electron density overlapping in hydrogen bonds: Topology vs. energetics. Chem. Phys. Lett. 1999, 300, 745–748. [Google Scholar] [CrossRef]
- Abramov, Y.A. On the possibility of kinetic energy density evaluation from the experimental electron-density distribution. Acta Cryst. A 1997, A53, 264–272. [Google Scholar] [CrossRef]
- Bogdan, E.; Compain, G.; Mtashobya, L.; Le Questel, J.-Y.; Besseau, F.; Galland, N.; Linclau, B.; Graton, J. Influence of fluorination on the conformational properties and hydrogen-bond acidity of benzyl alcohol derivatives. Chem. Eur. J. 2015, 21, 11462–11474. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, E.; Quarré de Verneuil, A.; Besseau, F.; Compain, G.; Linclau, B.; Le Questel, J.-Y.; Graton, J. α-fluoro-o-cresols: The key role of intramolecular hydrogen bonding in conformational preference and hydrogen-bond acidity. ChemPhysChem 2016, 17, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. Natural Bond Orbital (NBO) Version 6.0; Theoretical Chemistry Institute: Madison, WI, USA, 2013. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 3JH4,OH (Hz) | δOH4 (ppm) | h1JOH···F (Hz) | 3JH3,OH (Hz) | δOH3 (ppm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exp | Calc 1 | Exp | Calc 1 | Exp | Calc 1 | Exp | Calc 1 | Exp | Calc 1 | |
| 5 | 9.5 | 12.0 | 2.29 | 2.20 | - | - | 7.5 | 9.0 | 2.56 | 2.68 |
| 4 | 10.9 | 12.9 | 2.58 | 2.46 | 1.4 | 0.9 | 6.4 | 7.7 | 2.15 | 2.04 |
| 6 | 11.6 | 13.2 | 2.88 | 2.77 | v | - | 7.7 | 8.9 | 2.40 | 2.48 |
| 7 | 9.9 | 10.7 | 2.39 | 2.29 | - | - | - | - | - | - |
| 8 | 11.3 | 12.6 | 2.60 | 2.50 | 0.8 | 0.3 | - | - | - | - |
| 9 | - | - | - | - | - | - | 6.1 | 7.7 | 2.30 | 1.99 |
| Conformer 2 | G (kJ∙mol−1) | pi (%) | dOH4···O5 (Å) | dOH4···X2(ax) (Å) | HO4CH (°) | 3JOH4-H (Hz) | δOH4 (ppm) | |
|---|---|---|---|---|---|---|---|---|
| 5 | t_t | 0.0 | 65.2% | 2.377 | - | 165.0 | 13.6 | 2.31 |
| t_g | 3.8 | 14.0% | 2.324 | - | 164.2 | 13.5 | 2.49 | |
| t_g- | 5.4 | 7.5% | 2.319 | - | 163.7 | 13.2 | 2.53 | |
| g-_t | 5.5 | 7.0% | - | - | −56.7 | 3.0 | 1.31 | |
| g_t | 6.1 | 5.5% | - | - | 68.7 | 0.8 | 1.05 | |
| g_g- | 12.2 | 0.5% | - | - | 76.0 | −0.2 | 1.05 | |
| g-_g- | 13.4 | 0.3% | - | - | −60.2 | 2.8 | 1.24 | |
| 4 | t_t | 0.0 | 49.8% | 2.465 | 2.595 | 175.0 | 13.8 | 2.49 |
| t_g | 1.8 | 23.7% | 2.388 | 2.802 | 170.8 | 13.7 | 2.62 | |
| t_g- | 2.2 | 20.2% | 2.386 | 2.782 | 170.7 | 13.4 | 2.61 | |
| g-_t | 8.4 | 1.7% | - | - | −60.1 | 1.7 | 1.34 | |
| g_t | 8.8 | 1.4% | - | - | 65.3 | 0.9 | 1.08 | |
| g_g | 9.5 | 1.1% | - | - | 67.5 | 0.6 | 1.04 | |
| g-_g | 10.1 | 0.9% | - | - | −57.1 | 2.2 | 1.36 | |
| g_g- | 10.2 | 0.8% | - | - | −54.5 | −0.1 | 1.07 | |
| g-_g- | 11.5 | 0.5% | - | - | −58.2 | 1.2 | 1.29 | |
| 6 | t_t_g | 0.0 | 48.1% | 2.511 | 2.384 | −176.4 | 13.4 | 2.83 |
| t_t_g- | 1.8 | 23.7% | 2.539 | 2.324 | −174.1 | 13.3 | 2.88 | |
| t_g_g | 4.1 | 9.2% | 2.408 | 2.627 | 176.3 | 13.6 | 2.66 | |
| t_g-_g | 4.5 | 7.8% | 2.413 | 2.603 | 177.1 | 13.4 | 2.69 | |
| t_g_g- | 5.5 | 5.2% | 2.431 | 2.578 | 178.6 | 13.7 | 2.65 | |
| t_g-_g- | 5.7 | 4.8% | 2.425 | 2.588 | 178.0 | 13.4 | 2.65 | |
| g-_t_g | 11.4 | 0.5% | - | - | −64.1 | 1.1 | 1.14 | |
| g-_t_g- | 12.8 | 0.3% | - | - | −67.2 | 0.7 | 1.10 | |
| g_t_g- | 12.9 | 0.3% | - | - | 67.0 | 0.7 | 0.97 | |
| g_t_g | 13.3 | 0.2% | - | - | 67.1 | 0.6 | 0.93 | |
| 7 | t | 0.0 | 82.9% | 2.321 | - | 163.4 | 12.6 | 2.50 |
| g | 5.2 | 10.0% | - | - | 77.9 | −0.3 | 1.11 | |
| g- | 6.1 | 7.1% | - | - | −49.3 | 3.4 | 1.49 | |
| 8 | t | 0.0 | 92.6% | 2.390 | 2.840 | 169.5 | 13.5 | 2.60 |
| g | 7.6 | 4.3% | - | - | 68.9 | 0.6 | 1.18 | |
| g- | 8.4 | 3.1% | - | - | −52.6 | 2.9 | 1.54 | |
| Conformer 2 | G (kJ∙mol−1) | pi (%) | dOH3···O6 (Å) | HO3CH (°) | 3JOH3-H (Hz) | δOH3 (ppm) | |
|---|---|---|---|---|---|---|---|
| 5 | t_t | 0.0 | 65.2% | 2.122 | 156.9 | 11.0 | 3.15 |
| t_g | 3.8 | 14.0% | - | 81.6 | −0.2 | 0.74 | |
| t_g- | 5.4 | 7.5% | - | −50.8 | 3.9 | 1.33 | |
| g-_t | 5.5 | 7.0% | 2.109 | 158.2 | 11.1 | 3.34 | |
| g_t | 6.1 | 5.5% | 2.116 | 158.2 | 11.3 | 3.26 | |
| g_g- | 12.2 | 0.5% | - | 76.0 | 4.0 | 1.21 | |
| g-_g- | 13.4 | 0.3% | - | −60.2 | 3.5 | 1.20 | |
| 4 | t_t | 0.0 | 49.8% | 2.136 | 158.5 | 12.0 | 2.66 |
| t_g | 1.8 | 23.7% | - | 60.4 | 1.7 | 1.20 | |
| t_g- | 2.2 | 20.2% | - | −50.1 | 4.1 | 1.56 | |
| g-_t | 8.4 | 1.7% | 2.169 | 159.8 | 12.2 | 2.59 | |
| g_t | 8.8 | 1.4% | 2.185 | 160.6 | 12.6 | 2.47 | |
| g_g | 9.5 | 1.1% | - | 62.2 | 1.5 | 1.14 | |
| g-_g | 10.1 | 0.9% | - | 64.4 | 1.0 | 1.15 | |
| g_g- | 10.2 | 0.8% | - | −54.5 | 3.2 | 1.39 | |
| g-_g- | 11.5 | 0.5% | - | −58.2 | 2.6 | 1.36 | |
| 6 | t_t_g | 0.0 | 48.1% | 2.094 | 158.4 | 11.5 | 2.97 |
| t_t_g- | 1.8 | 23.7% | 2.078 | 157.6 | 11.7 | 3.07 | |
| t_g_g | 4.1 | 9.2% | - | 77.1 | −0.3 | 0.77 | |
| t_g-_g | 4.5 | 7.8% | - | −52.2 | 3.2 | 1.35 | |
| t_g_g- | 5.5 | 5.2% | - | 68.8 | 0.5 | 0.98 | |
| t_g-_g- | 5.7 | 4.8% | - | −51.8 | 3.4 | 1.34 | |
| g-_t_g | 11.4 | 0.5% | 2.173 | 159.7 | 11.8 | 2.64 | |
| g-_t_g- | 12.8 | 0.3% | 2.138 | 159.0 | 11.9 | 2.80 | |
| g_t_g | 12.9 | 0.3% | 2.173 | 160.0 | 12.0 | 2.59 | |
| g_t_g- | 13.3 | 0.2% | 2.157 | 159.8 | 12.3 | 2.66 | |
| 9 | t | 0.0 | 51.0% | 2.186 | 160.3 | 12.4 | 2.50 |
| g- | 1.8 | 24.9% | - | −48.8 | 4.3 | 1.64 | |
| g | 1.9 | 24.1% | - | 63.4 | 1.5 | 1.28 | |
| Conformer | E(2)n→σ* nO6 → σ*OH3 (kJ∙mol−1) | ρOH3···O6 e∙bohr−3 | EHB (kJ∙mol−1) | E(2)n→σ* nO5 → σ*OH4 (kJ∙mol−1) | E(2)n→σ* nX2 → σ*OH4 (kJ∙mol−1) | ρOH4···OMe e∙bohr−3 | EHB (kJ∙mol−1) | |
|---|---|---|---|---|---|---|---|---|
| 4 | t_t | 11.9 | 0.020 | 20.0 | 1.6 | 0.4 | - | - |
| t_g | - | - | - | 2.4 | 0.2 | - | - | |
| t_g- | - | - | - | 2.4 | 0.3 | - | - | |
| g-_t | 10.5 | 0.018 | 18.5 | - | - | - | - | |
| g_t | 9.7 | 0.018 | 17.9 | - | - | - | - | |
| 5 | t_t | 12.6 | 0.020 | 20.5 | 2.8 | - | - | - |
| t_g | - | - | - | 3.5 | - | - | - | |
| t_g- | - | - | - | 3.6 | - | - | - | |
| g-_t | 13.6 | 0.021 | 21.1 | - | - | - | - | |
| g_t | 13.1 | 0.020 | 20.7 | - | - | - | - | |
| 6 | t_t_g | 14.1 | 0.021 | 22.1 | 1.2 | 3.8 | 0.011 | 10.5 |
| t_t_g- | 15.0 | 0.022 | 23.0 | 1.0 | 4.2 | 0.012 | 11.4 | |
| t_g_g | - | - | - | 2.1 | 0.8 | 0.007 | 7.0 | |
| t_g-_g | - | - | - | 2.1 | 1.0 | 0.008 | 7.2 | |
| t_g_g- | - | - | - | 1.9 | 1.1 | 0.008 | 7.2 | |
| t_g-_g- | - | - | - | 2.0 | 1.1 | 0.007 | 7.1 | |
| 7 | t | - | - | - | 2.4 | 0.08 | - | - |
| 8 | t | - | - | - | 3.6 | - | - | - |
| 9 | t | 9.7 | 0.018 | 17.9 | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiquempoix, L.; Bogdan, E.; Wells, N.J.; Le Questel, J.-Y.; Graton, J.; Linclau, B. A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives. Molecules 2017, 22, 518. https://doi.org/10.3390/molecules22040518
Quiquempoix L, Bogdan E, Wells NJ, Le Questel J-Y, Graton J, Linclau B. A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives. Molecules. 2017; 22(4):518. https://doi.org/10.3390/molecules22040518
Chicago/Turabian StyleQuiquempoix, Lucas, Elena Bogdan, Neil J. Wells, Jean-Yves Le Questel, Jérôme Graton, and Bruno Linclau. 2017. "A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives" Molecules 22, no. 4: 518. https://doi.org/10.3390/molecules22040518
APA StyleQuiquempoix, L., Bogdan, E., Wells, N. J., Le Questel, J. -Y., Graton, J., & Linclau, B. (2017). A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives. Molecules, 22(4), 518. https://doi.org/10.3390/molecules22040518