
Optimizing Sample Size to Assess the Genetic Diversity in Common Vetch (Vicia sativa L.) Populations Using Start Codon Targeted (SCoT) Markers



Abstract
:1. Introduction
2. Results
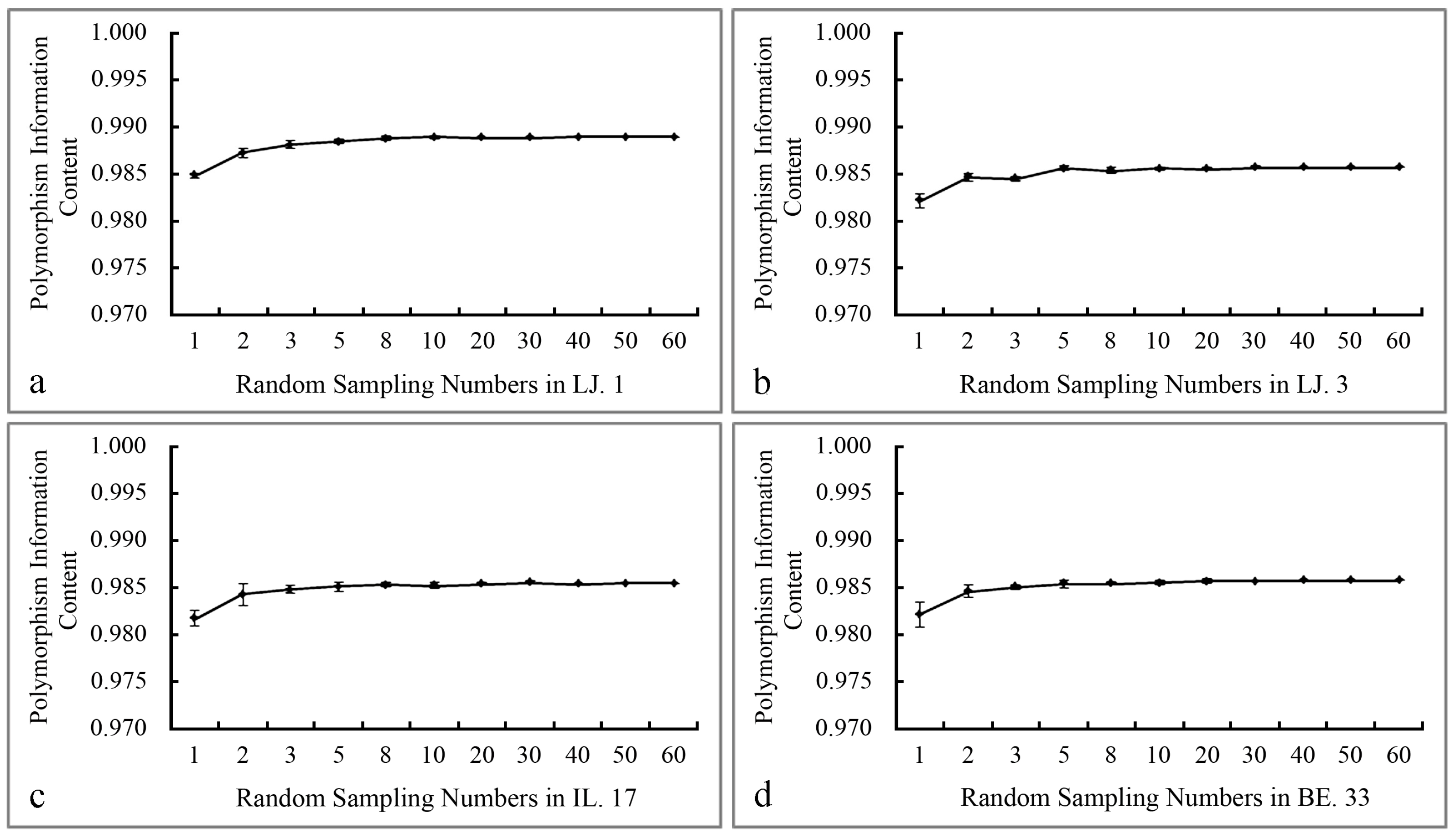
2.1. The Polymorphism and Genetic Diversity Analysis of SCoT Markers
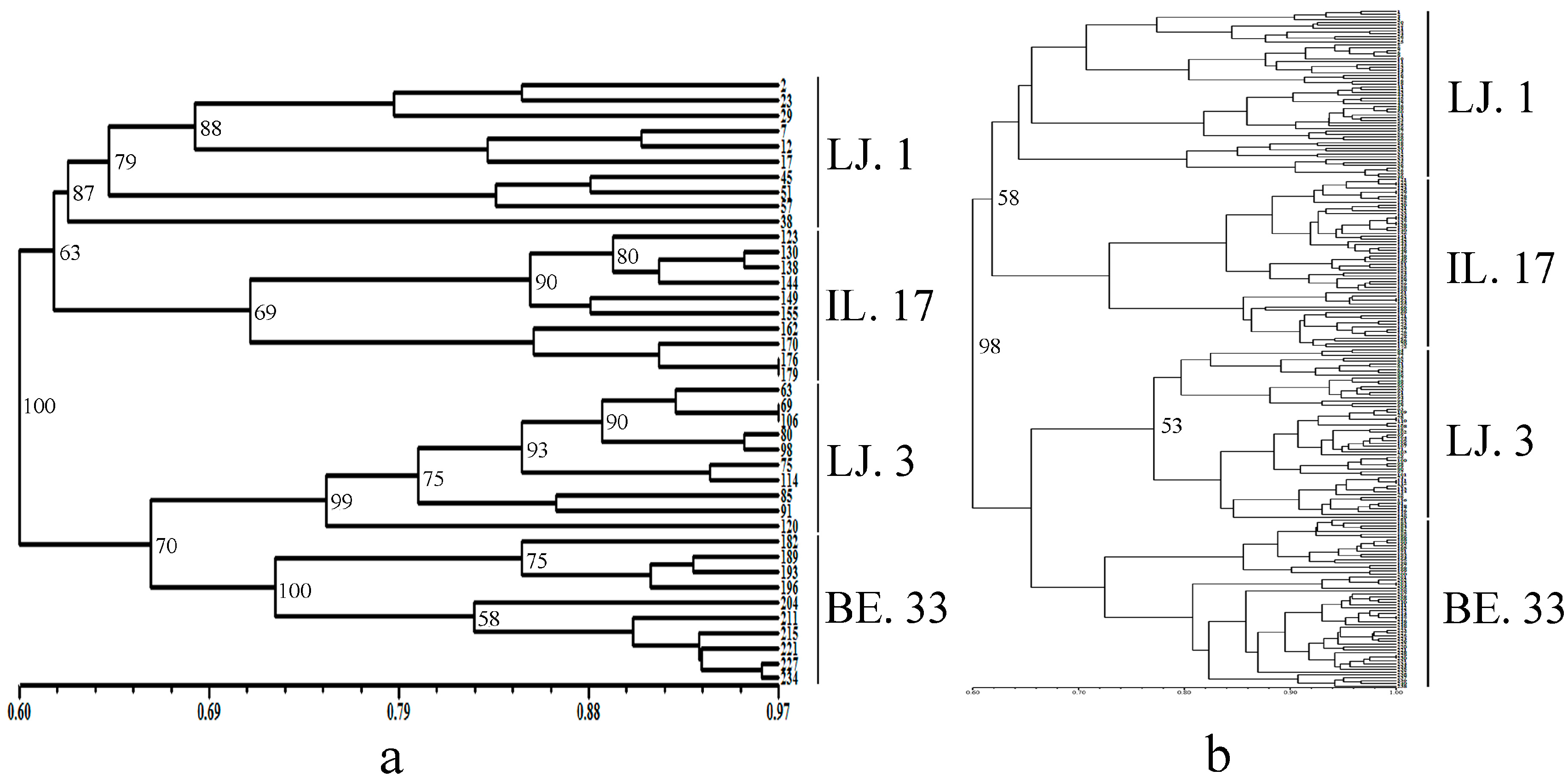
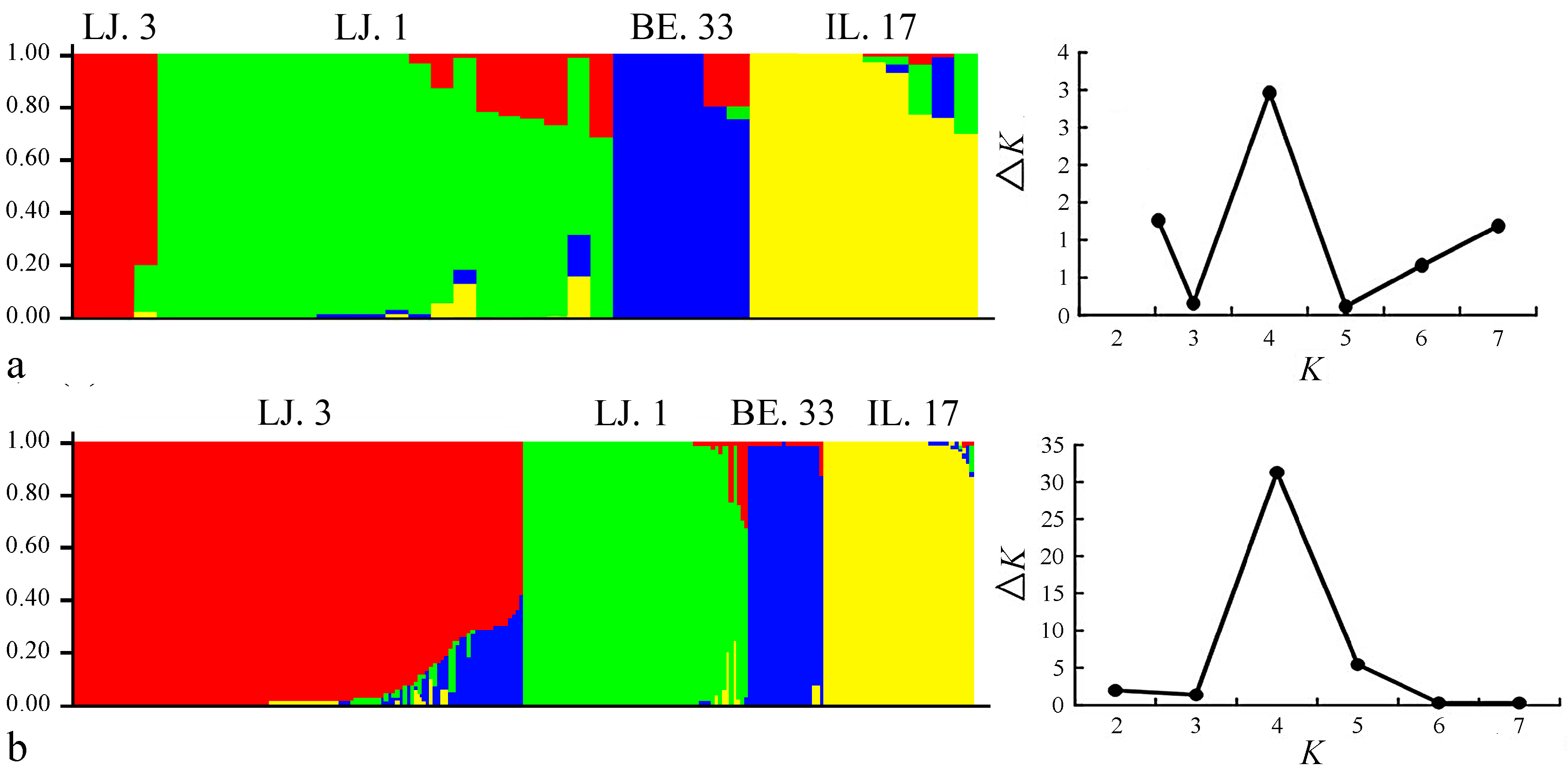
2.2. Cluster and Population Structure Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. PCR Amplification
4.3. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arslan, E.; Ertuğrul, K.; Öztürk, A.B. Karyological studies of some species of the genus Vicia L. (Leguminosae) in Turkey. Caryologia 2012, 65, 106–113. [Google Scholar] [CrossRef]
- Liu, Z.P.; Liu, P.; Luo, D.; Liu, W.X.; Wang, Y.R. Exploiting Illumina sequencing for the development of 95 novel polymorphic EST-SSR markers in common vetch (Vicia sativa subsp. sativa). Molecules 2014, 19, 5777–5789. [Google Scholar] [CrossRef] [PubMed]
- Potokina, E.; Blattner, F.; Alexandrova, T.; Bachmann, K. AFLP diversity in the common vetch (Vicia sativa L.) on the world scale. Theor. Appl. Genet. 2002, 105, 58–67. [Google Scholar] [PubMed]
- Chung, J.W.; Kim, T.S.; Suresh, S.; Lee, S.Y.; Cho, G.T. Development of 65 novel polymorphic cDNA-SSR markers in common vetch (Vicia sativa subsp. sativa) using next generation sequencing. Molecules 2013, 18, 8376–8392. [Google Scholar] [PubMed]
- Chung, J.W.; Kim, T.S.; Sundan, S.; Lee, G.A.; Park, J.H.; Cho, G.T.; Lee, H.S.; Lee, J.Y.; Lee, M.C.; Baek, H.J.; et al. New cDNA-SSR markers in the narrow-leaved vetch (Vicia sativa subsp. nigra) using 454 pyrosequencing. Mol. Breed. 2014, 33, 749–754. [Google Scholar] [CrossRef]
- Kim, T.S.; Raveendar, S.; Suresh, S.; Lee, G.A.; Lee, J.R.; Cho, J.H.; Lee, S.Y.; Ma, K.H.; Cho, G.T.; Chung, J.W. Transcriptome analysis of two Vicia sativa subspecies: Mining molecular markers to enhance genomic resources for vetch improvement. Genes 2015, 6, 1164–1182. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, J.M.; Kelly, J.D. Agronomic traits affecting resistance to white mold in common bean. Crop Sci. 2002, 42, 693–699. [Google Scholar] [CrossRef]
- Dhima, K.V.; Lithourgidis, A.S.; Vasilakoglou, I.B.; Dordas, C.A. Competition indices of common vetch and cereal intercrops in two seeding ratio. Field Crop. Res. 2007, 100, 249–256. [Google Scholar] [CrossRef]
- Kahlaoui, S.; Walker, D.J.; Correal, E.; Martínez-Gómez, P.; Hassen, H.; Bouzid, S. The morphology, chromosome number and nuclear DNA content of Tunisian populations of three Vicia species. Afr. J. Biotechnol. 2009, 8, 3184–3191. [Google Scholar]
- Jaaska, V. Isozyme evidence on the specific distinctness and phylogenetic position of Vicia incise (Fabaceae). Open Life Sci. 2008, 3, 169–176. [Google Scholar]
- Arias, M.; Hernandez, M.; Remondegui, N.; Huvenaars, K.; Van, D.P.; Ritter, E. First genetic linkage map of Taraxacum koksaghyz Rodin based on AFLP, SSR, COS and EST-SSR markers. Sci. Rep. 2016, 6, 31031. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, M.; Hodkinson, T.R.; Barth, S. Chloroplast DNA markers (cpSSRs, SNPs) for Miscanthus, Saccharum and related grasses (Panicoideae, Poaceae). Mol. Breed. 2010, 26, 539–544. [Google Scholar] [CrossRef]
- Lespinasse, D.; Rodiergoud, M.; Grivet, L.; Leconte, A.; Legnate, H.; Seguin, M. A saturated genetic linkage map of rubber tree (Hevea spp.) based on RFLP, AFLP, microsatellite, and isozyme markers. Theor. Appl. Genet. 2000, 100, 127–138. [Google Scholar] [CrossRef]
- Liu, Z.P.; Chen, T.L.; Ma, L.C.; Zhao, Z.G.; Zhao, P.X.; Nan, Z.B.; Wang, Y.R. Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS ONE 2013, 8, 83549. [Google Scholar]
- Zhou, Q.; Luo, D.; Ma, L.C.; Xie, W.G.; Wang, Y.R.; Liu, Z.P. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6, 20549. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, P.; Kumaria, S.; Kumar, S.; Tandon, P. Start Codon Targeted (SCoT) marker reveals genetic diversity of Dendrobium nobile Lindl., an endangered medicinal orchid species. Gene 2013, 529, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.Y.; Mackill, D.J. Start codon targeted (SCoT) polymorphism: A simple, novel DNA marker technique for generating gene-targeted markers in plants. Plant Mol. Biol. Rep. 2008, 27, 86–93. [Google Scholar] [CrossRef]
- Guo, D.L.; Zhang, J.Y.; Liu, C.H. Genetic diversity in some grape varieties revealed by SCoT analyses. Mol. Biol. Rep. 2012, 39, 5307–5313. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; He, X.H.; Chen, H.; Ou, S.J.; Gao, M.P. Analysis of diversity and relationships among mango cultivars using Start Codon Targeted (SCoT) markers. Biochem. Syst. Ecol. 2010, 38, 1176–1184. [Google Scholar] [CrossRef]
- Xiong, F.; Zhong, R.C.; Han, Z.Q.; Jiang, J.; He, L.Q.; Zhuang, W.J.; Tang, R.H. Start codon targeted polymorphism for evaluation of functional genetic variation and relationships in cultivated peanut (Arachishypogaea L.) genotypes. Mol. Biol. Rep. 2011, 38, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Xie, W.G.; Wang, Y.R.; Zhao, X.H. Potential of start codon targeted (SCoT) markers to estimate genetic diversity and relationships among chinese Elymus sibiricus accessions. Molecules 2015, 20, 5987–6001. [Google Scholar] [CrossRef] [PubMed]
- Pakseresht, F.; Talebi, R.; Karami, E. Comparative assessment of ISSR, DAMD and SCoT markers for evaluation of genetic diversity and conservation of landrace chickpea (Cicer arietinum L.) genotypes collected from north-west of Iran. Physiol. Mol. Biol. Plants 2013, 19, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wang, M.; Zeng, J.; Yan, W.M.; Wang, J.Q.; Ma, T.; Hang, R. Bacterial community compositions in response to sediment properties in urban lakes of Nanjing. Afr. J. Microbiol. Res. 2012, 6, 2929–2940. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Engelmann, F. Use of biotechnologies for the conservation of plant biodiversity. In Vitro Cell. Dev. Plant 2011, 47, 5–16. [Google Scholar] [CrossRef]
- Sjögren, P.; Wyöni, P.I. Conservation genetics and detection of rare alleles in finite populations. Conserv. Biol. 1994, 8, 267–270. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Zhou, T.Y.; Zhong, M.; Lu, B.R. Sampling strategy for wild soybean (Glycine soja) populations based on their genetic diversity and fine-scale spatial genetic structure. Front. Biol. 2007, 2, 397–402. [Google Scholar] [CrossRef]
- Che, Y.H.; Li, L.H.; He, P.R. Sampling size for genetic diversity in Agropyron Gaertn. based on gliadin. J. Plant Genet. Resour. 2004, 3, 216–221. [Google Scholar]
- Jiang, L.F.; Qi, X.; Zhang, X.Q.; Huang, L.K.; Ma, X.; Xie, W.G. Analysis of diversity and relationships among orchardgrass (Dactylis glomerata L.) accessions using start codon-targeted markers. Genet. Mol. Res. 2014, 13, 4406–4418. [Google Scholar] [CrossRef] [PubMed]
- Gajera, H.P.; Hirpara, D.G.; Katakpara, Z.A.; Patel, S.V.; Golakiya, B.A. Molecular evolution and phylogenetic analysis of biocontrol genes acquired from SCoT polymorphism of mycoparasitic Trichoderma koningii inhibiting phytopathogen Rhizoctonia solani Kuhn. Infect. Genet. Evol. 2016, 45, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.W.; Pan, J.; Min, D.D.; Fan, Y.; Ding, X.Y.; Fan, S.G.; Baskin, C.C.; Baskin, J.M. Seed dormancy and soil seedbank of the invasive weed Chenopodium hybridum in north-western china. Weed Res. 2016, 57, 54–64. [Google Scholar] [CrossRef]
- Gawel, N.J.; Jarret, R.L. A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Mol. Biol. Rep. 1991, 9, 262–266. [Google Scholar] [CrossRef]
- Borovics, A.; Mátyás, C. Decline of genetic diversity of sessile oak at the retracting (xeric) limits. Ann. For. Sci. 2013, 70, 835–844. [Google Scholar] [CrossRef]
- Huang, S.P.; Weir, B.S. Estimating the total number of alleles using a sample coverage method. Genetics 2001, 159, 1365–1373. [Google Scholar] [PubMed]
- Liu, W.X.; Li, L.H.; Liu, W.H. SSR analysis on the sampling strategy of Psathyrostachys huashanic Keng population. J. Triticeae Crops 2006, 26, 16–20. [Google Scholar]
- Fitzpatrick, B.M. Power and sample size for nested analysis of molecular variance. Mol. Ecol. 2009, 18, 3961–3966. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System; Exeter Software: New York, NY, USA, 1992. [Google Scholar]
- Pavlicek, A.; Hrda, S.; Flegr, J. FreeTree-freeware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness. Application in the RAPD analysis of genus Frenkelia. Folia Biol. 1999, 45, 97–99. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar]
- Prevost, A.; Wilkinson, M. A new system of comparing PCR primers applied to ISSR fingerprinting of potato cultivars. Theor. Appl. Genet. 1999, 98, 107–112. [Google Scholar] [CrossRef]
- Yeh, F.; Yang, R.; Boyle, T. Quick User Guide. In Popgene; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Accessions | Type of Accessions | Sample Code | Origin of Germplasm |
|---|---|---|---|
| LJ. 1 | Cultivated variety, Lanjian No. 1 | 1–60 | China |
| LJ. 3 | Cultivated variety, Lanjian No. 3 | 61–120 | China |
| IL. 17 | Wild accession, No. 17 | 121–180 | Israel |
| BE. 33 | Wild accession, No. 33 | 181–240 | Belgium |
| Sampling No. (Sampling Size) | LJ. 1 | LJ. 3 | IL. 17 | BE. 33 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Na | RP | HE | Na | RP | HE | Na | RP | HE | Na | RP | HE | |
| 1 (1) | 67 | 58% | 0.9851 | 59 | 56% | 0.9823 | 56 | 57% | 0.9822 | 58 | 55% | 0.9825 |
| 2 (2) | 87 | 75% | 0.9874 | 72 | 69% | 0.9847 | 71 | 72% | 0.9846 | 72 | 69% | 0.9849 |
| 3 (3) | 97 | 84% | 0.9882 | 74 | 70% | 0.9845 | 79 | 80% | 0.9851 | 79 | 75% | 0.9853 |
| 4 (5) | 104 | 89% | 0.9886 | 84 | 80% | 0.9856 | 87 | 88% | 0.9854 | 85 | 81% | 0.9857 |
| 5 (8) | 108 | 93% | 0.9889 | 88 | 84% | 0.9854 | 88 | 89% | 0.9856 | 91 | 87% | 0.9857 |
| 6 (10) | 112 | 97% | 0.9890 | 95 | 90% | 0.9856 | 91 | 92% | 0.9856 | 94 | 90% | 0.9858 |
| 7 (20) | 113 | 97% | 0.9889 | 96 | 91% | 0.9855 | 95 | 96% | 0.9857 | 98 | 93% | 0.9860 |
| 8 (30) | 114 | 98% | 0.9890 | 100 | 95% | 0.9857 | 97 | 98% | 0.9858 | 100 | 95% | 0.9859 |
| 9 (40) | 115 | 99% | 0.9890 | 102 | 97% | 0.9857 | 98 | 99% | 0.9857 | 104 | 99% | 0.9860 |
| 10 (50) | 116 | 100% | 0.9890 | 103 | 98% | 0.9857 | 99 | 100% | 0.9858 | 104 | 99% | 0.9860 |
| 11 (60) | 116 | 100% | 0.9890 | 105 | 100% | 0.9857 | 99 | 100% | 0.9858 | 105 | 100% | 0.9860 |
| Sampling No. (Sampling Size) | LJ. 1 | LJ. 3 | IL. 17 | BE. 33 |
|---|---|---|---|---|
| 1 (1) | 0.00 | 0.00 | 0.00 | 0.00 |
| 2 (2) | 0.25 | 0.31 | 0.33 | 0.34 |
| 3 (3) | 0.34 | 0.37 | 0.36 | 0.37 |
| 4 (5) | 0.35 | 0.40 | 0.39 | 0.39 |
| 5 (8) | 0.39 | 0.41 | 0.42 | 0.42 |
| 6 (10) | 0.44 | 0.44 | 0.44 | 0.45 |
| 7 (20) | 0.44 | 0.43 | 0.44 | 0.45 |
| 8 (30) | 0.43 | 0.44 | 0.44 | 0.44 |
| 9 (40) | 0.44 | 0.45 | 0.45 | 0.45 |
| 10 (50) | 0.45 | 0.45 | 0.45 | 0.45 |
| 11 (60) | 0.45 | 0.45 | 0.45 | 0.45 |
| Shannon information index | 0.67 | 0.68 | 0.69 | 0.69 |
| Primer | LJ. 1 | LJ. 3 | IL. 17 | BE. 33 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rp | HE | PIC | Rp | HE | PIC | Rp | HE | PIC | Rp | HE | PIC | |
| Scot28 | 8.53 | 0.9442 | 0.9413 | 5.90 | 0.9282 | 0.9235 | 3.07 | 0.8834 | 0.8720 | 6.00 | 0.9114 | 0.9046 |
| Scot35 | 13.67 | 0.9413 | 0.9381 | 6.40 | 0.9216 | 0.9160 | 10.73 | 0.9344 | 0.9305 | 11.10 | 0.9345 | 0.9306 |
| Scot36 | 13.90 | 0.9518 | 0.9496 | 7.50 | 0.9194 | 0.9138 | 4.20 | 0.9374 | 0.9337 | 7.43 | 0.9216 | 0.9161 |
| Scot37 | 9.17 | 0.9408 | 0.9376 | 4.33 | 0.9248 | 0.9197 | 9.10 | 0.9275 | 0.9227 | 6.10 | 0.9309 | 0.9266 |
| Scot38 | 8.63 | 0.9511 | 0.9488 | 5.87 | 0.9442 | 0.9412 | 6.03 | 0.9445 | 0.9416 | 2.67 | 0.9492 | 0.9467 |
| Primer | Primer Sequence (5′-3′) | Primer | Primer Sequence (5′-3′) |
|---|---|---|---|
| Scot12 | ACGACATGGCGACCAACG | Scot41 | CAATGGCTACCACTGACA |
| Scot13 | ACGACATGGCGACCATCG | Scot42 | CAATGGCTACCATTAGCG |
| Scot14 | ACGACATGGCGACCACGC | Scot43 | CAATGGCTACCACCGCAG |
| Scot15 | ACGACATGGCGACCGCGA | Scot44 | CAATGGCTACCATTAGCC |
| Scot16 | ACCATGGCTACCACCGAC | Scot45 | ACAATGGCTACCACTGAC |
| Scot23 | CACCATGGCTACCACCAG | Scot46 | ACAATGGCTACCACTGAG |
| Scot28 | CCATGGCTACCACCGCCA | Scot47 | ACAATGGCTACCACTGCC |
| Scot35 | CATGGCTACCACCGGCCC | Scot48 | ACAATGGCTACCACTGGC |
| Scot36 | GCAACAATGGCTACCACC | Scot49 | ACAATGGCTACCACTGCG |
| Scot37 | CAATGGCTACCACTAGCC | Scot50 | ACAATGGCTACCACTGGG |
| Scot38 | CAATGGCTACCACTAACG | Scot51 | ACAATGGCTACCACTGTC |
| Scot39 | CAATGGCTACCACTAGCG | Scot52 | ACAATGGCTACCACTGCA |
| Scot40 | CAATGGCTACCACTACAG |
| Primer | Tm/°C | PPB | Na | HE | PIC | Range of Band Size (bp) |
|---|---|---|---|---|---|---|
| Scot28 | 61.9 | 100% | 25 | 0.9374 | 0.9339 | 200–2100 |
| Scot35 | 64.1 | 100% | 21 | 0.9429 | 0.9400 | 200–1900 |
| Scot36 | 57.3 | 100% | 29 | 0.9540 | 0.9520 | 240–2200 |
| Scot37 | 61.9 | 100% | 23 | 0.9444 | 0.9415 | 250–2400 |
| Scot38 | 61.9 | 100% | 24 | 0.9518 | 0.9496 | 220–1800 |
| Average | 100% | 24.4 | 0.9461 | 0.9434 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, X.; Dong, R.; Liu, W.; Wang, Y.; Liu, Z. Optimizing Sample Size to Assess the Genetic Diversity in Common Vetch (Vicia sativa L.) Populations Using Start Codon Targeted (SCoT) Markers. Molecules 2017, 22, 567. https://doi.org/10.3390/molecules22040567
Chai X, Dong R, Liu W, Wang Y, Liu Z. Optimizing Sample Size to Assess the Genetic Diversity in Common Vetch (Vicia sativa L.) Populations Using Start Codon Targeted (SCoT) Markers. Molecules. 2017; 22(4):567. https://doi.org/10.3390/molecules22040567
Chicago/Turabian StyleChai, Xutian, Rui Dong, Wenxian Liu, Yanrong Wang, and Zhipeng Liu. 2017. "Optimizing Sample Size to Assess the Genetic Diversity in Common Vetch (Vicia sativa L.) Populations Using Start Codon Targeted (SCoT) Markers" Molecules 22, no. 4: 567. https://doi.org/10.3390/molecules22040567
APA StyleChai, X., Dong, R., Liu, W., Wang, Y., & Liu, Z. (2017). Optimizing Sample Size to Assess the Genetic Diversity in Common Vetch (Vicia sativa L.) Populations Using Start Codon Targeted (SCoT) Markers. Molecules, 22(4), 567. https://doi.org/10.3390/molecules22040567