
Structural and Biochemical Properties of Novel Self-Cleaving Ribozymes

Abstract
:1. Introduction
2. Structures of the Novel Ribozymes
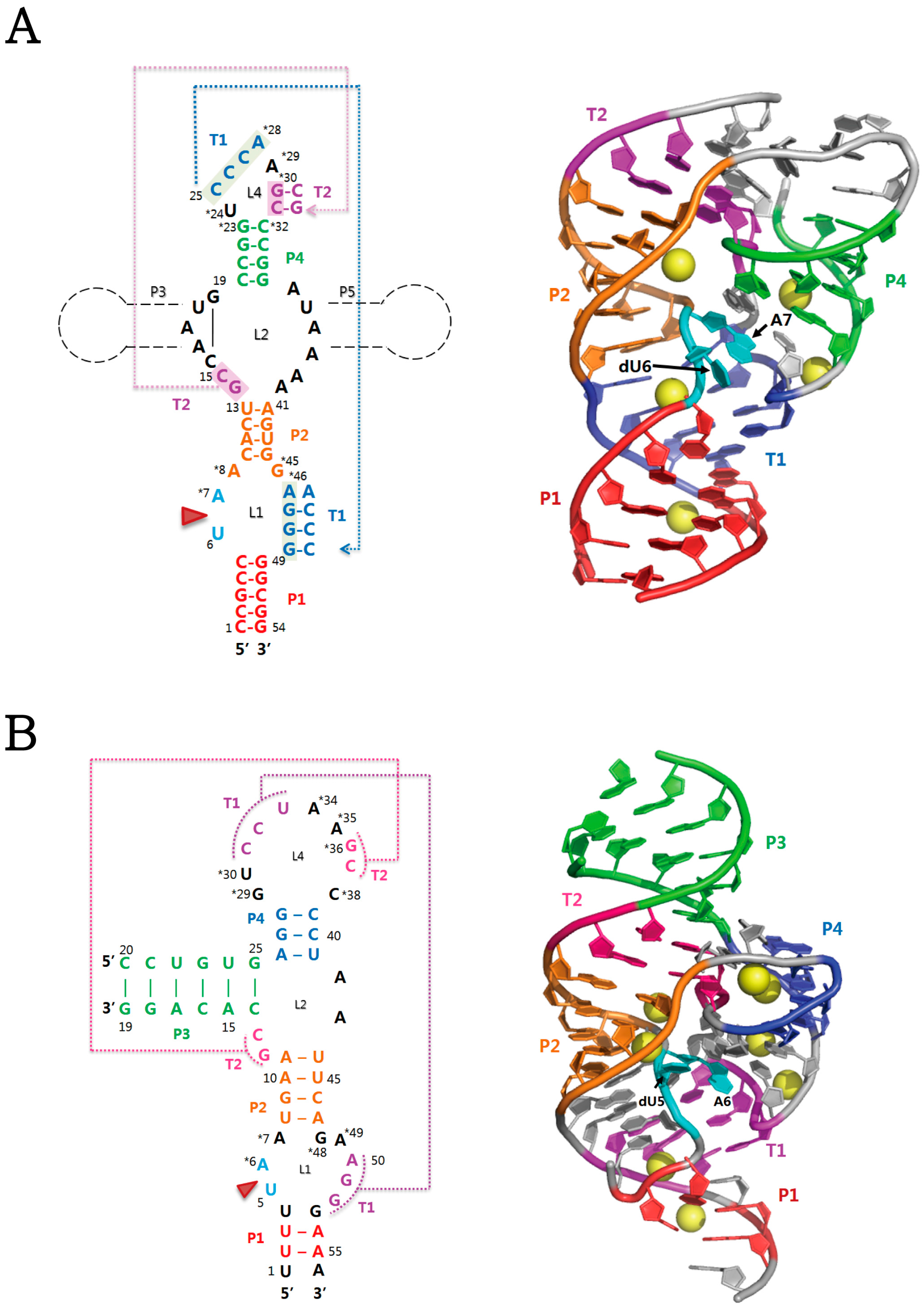
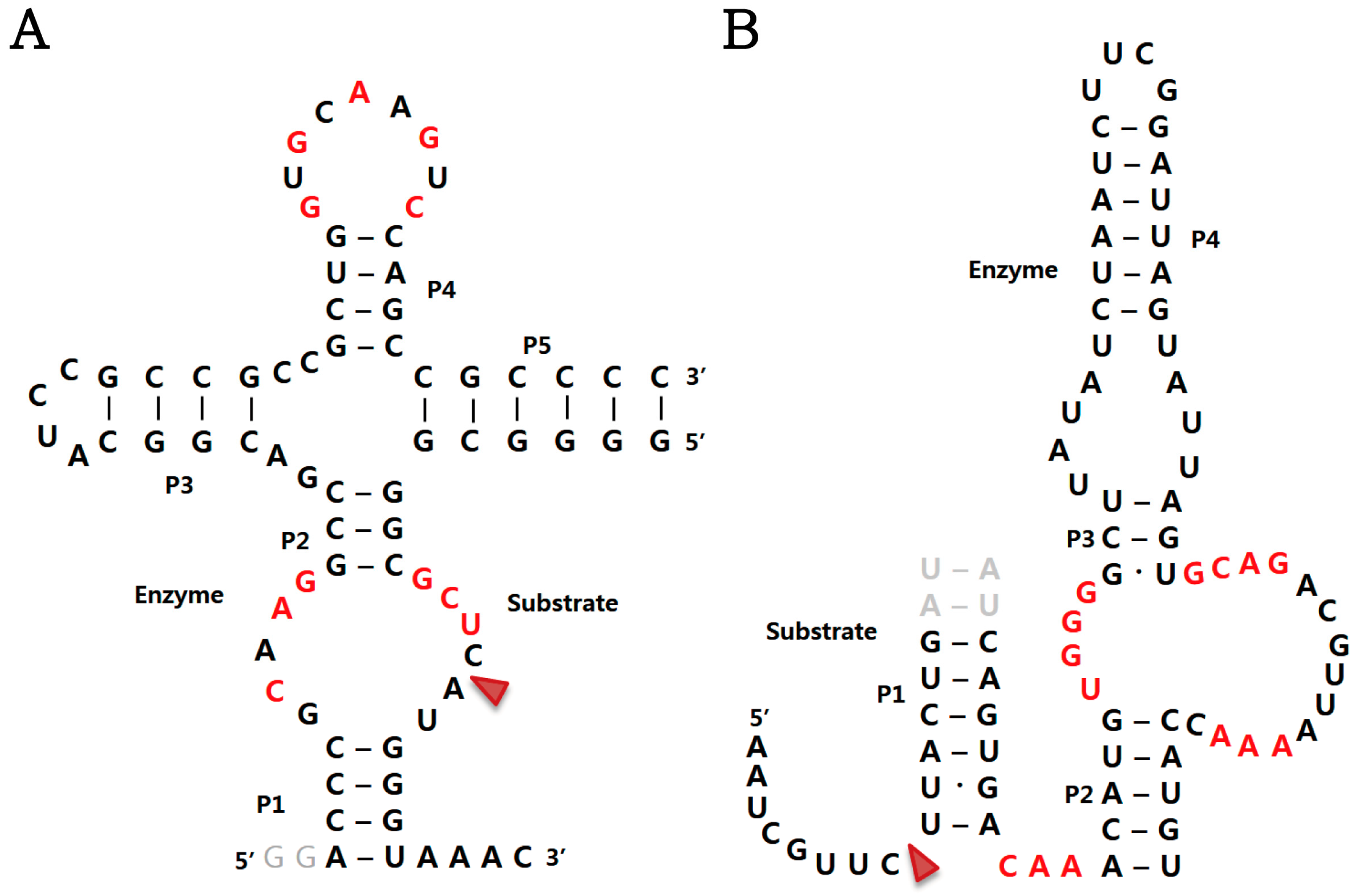
2.1. Twister Ribozyme
2.2. Twister Sister Ribozyme
2.3. Pistol Ribozyme
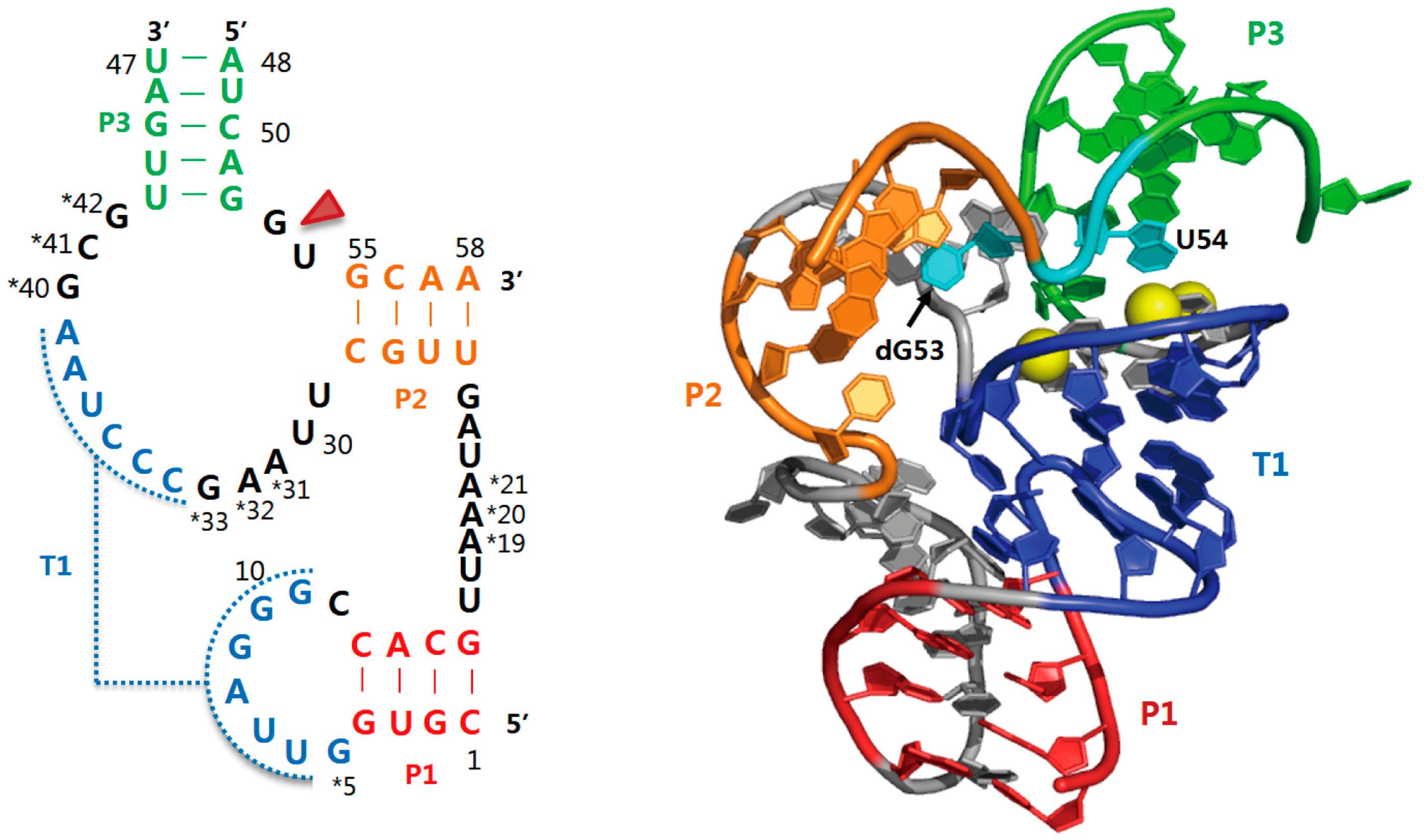
2.4. Hatchet Ribozyme
3. Insight into Catalytic Mechanisms of the Novel Ribozymes
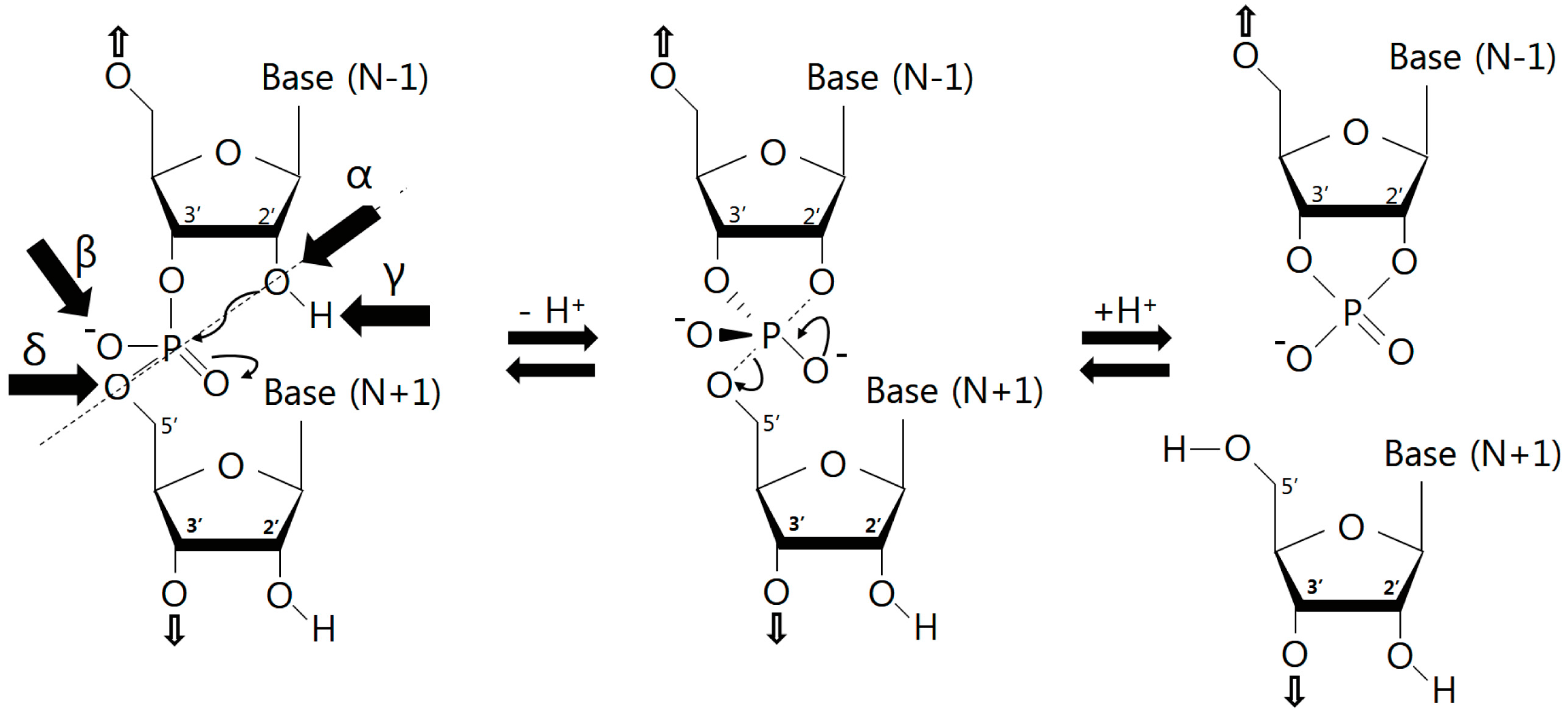
3.1. Catalytic Strategies
3.2. Nucleobase Involvement in Catalysis
3.3. Metal Ion-Dependent Catalysis
4. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.P.; Joyce, G.F. The origins of the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, a003608. [Google Scholar] [CrossRef] [PubMed]
- Prody, G.A.; Bakos, J.T.; Buzayan, J.M.; Schneider, I.R.; Bruening, G. Autolytic processing of dimeric plant virus satellite RNA. Science 1986, 231, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNAs of a virusoid and a structural model for the active sites. Cell 1987, 49, 211–220. [Google Scholar] [CrossRef]
- Muller, S.; Appel, B.; Balke, D.; Hieronymus, R.; Nubel, C. Thirty-five years of research into ribozymes and nucleic acid catalysis: Where do we stand today? F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, R.M.; Polanco, J.A.; Luptak, A. Chemistry and Biology of Self-Cleaving Ribozymes. Trends Biochem. Sci. 2015, 40, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Ferre-D’Amare, A.R.; Scott, W.G. Small self-cleaving ribozymes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003574. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Weinberg, Z.; Chen, A.G.; Kim, P.B.; Ames, T.D.; Breaker, R.R. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat. Chem. Biol. 2014, 10, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.; Kim, P.B.; Chen, T.H.; Li, S.; Harris, K.A.; Lunse, C.E.; Breaker, R.R. New classes of self-cleaving ribozymes revealed by comparative genomics analysis. Nat. Chem. Biol. 2015, 11, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.A.; Lunse, C.E.; Li, S.; Brewer, K.I.; Breaker, R.R. Biochemical analysis of pistol self-cleaving ribozymes. RNA 2015, 21, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lunse, C.E.; Harris, K.A.; Breaker, R.R. Biochemical analysis of hatchet self-cleaving ribozymes. RNA 2015, 21, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Fica, S.M.; Tuttle, N.; Novak, T.; Li, N.S.; Lu, J.; Koodathingal, P.; Dai, Q.; Staley, J.P.; Piccirilli, J.A. RNA catalyses nuclear pre-mRNA splicing. Nature 2013, 503, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, C.J.; Rathjen, P.D.; Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNA transcripts of avocado sunblotch viroid. Nucleic Acids Res. 1986, 14, 3627–3640. [Google Scholar] [CrossRef] [PubMed]
- Eickbush, D.G.; Eickbush, T.H. R2 retrotransposons encode a self-cleaving ribozyme for processing from an rRNA cotranscript. Mol. Cell. Biol. 2010, 30, 3142–3150. [Google Scholar] [CrossRef] [PubMed]
- Ruminski, D.J.; Webb, C.H.; Riccitelli, N.J.; Luptak, A. Processing and translation initiation of non-long terminal repeat retrotransposons by hepatitis delta virus (HDV)-like self-cleaving ribozymes. J. Biol. Chem. 2011, 286, 41286–41295. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Luque, F.J.; Lopez, M.C.; Macias, F.; Alonso, C.; Thomas, M.C. Identification of an hepatitis delta virus-like ribozyme at the mRNA 5’-end of the L1Tc retrotransposon from Trypanosoma cruzi. Nucleic Acids Res. 2011, 39, 8065–8077. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Nahvi, A.; Roth, A.; Collins, J.A.; Breaker, R.R. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004, 428, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Fedor, M.J. Structure and function of the hairpin ribozyme. J. Mol. Biol. 2000, 297, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Perreault, J.; Weinberg, Z.; Roth, A.; Popescu, O.; Chartrand, P.; Ferbeyre, G.; Breaker, R.R. Identification of hammerhead ribozymes in all domains of life reveals novel structural variations. PLoS Comput. Biol. 2011, 7, e1002031. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.H.; Riccitelli, N.J.; Ruminski, D.J.; Luptak, A. Widespread occurrence of self-cleaving ribozymes. Science 2009, 326, 953. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.Y.; Fedor, M.J. The ydaO motif is an ATP-sensing riboswitch in Bacillus subtilis. Nat. Chem. Biol. 2012, 8, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Lilley, D.M. The Varkud satellite ribozyme. RNA 2004, 10, 151–158. [Google Scholar] [CrossRef] [PubMed]
- McCown, P.J.; Roth, A.; Breaker, R.R. An expanded collection and refined consensus model of glmS ribozymes. RNA 2011, 17, 728–736. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.J.; Plog, M.A.; Floy, S.A.; Jansen, J.A.; Soukup, J.K.; Soukup, G.A. Ligand requirements for glmS ribozyme self-cleavage. Chem. Biol. 2005, 12, 1221–1226. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Ferre-D’Amare, A.R. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 2006, 313, 1752–1756. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.A.; Irnov, I.; Baker, S.; Winkler, W.C. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 2007, 21, 3356–3368. [Google Scholar] [CrossRef] [PubMed]
- Eiler, D.; Wang, J.; Steitz, T.A. Structural basis for the fast self-cleavage reaction catalyzed by the twister ribozyme. Proc. Natl. Acad. Sci. USA 2014, 111, 13028–13033. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Kosutic, M.; Rajashankar, K.R.; Frener, M.; Santner, T.; Westhof, E.; Micura, R.; Patel, D.J. In-line alignment and Mg2+ coordination at the cleavage site of the env22 twister ribozyme. Nat. Commun. 2014, 5, 5534. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wilson, T.J.; McPhee, S.A.; Lilley, D.M. Crystal structure and mechanistic investigation of the twister ribozyme. Nat. Chem. Biol. 2014, 10, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Kosutic, M.; Neuner, S.; Ren, A.; Flur, S.; Wunderlich, C.; Mairhofer, E.; Vusurovic, N.; Seikowski, J.; Breuker, K.; Hobartner, C.; et al. A Mini-Twister Variant and Impact of Residues/Cations on the Phosphodiester Cleavage of this Ribozyme Class. Angew. Chem. Int. Ed. Engl. 2015, 54, 15128–15133. [Google Scholar] [CrossRef] [PubMed]
- Pleij, C.W.; Rietveld, K.; Bosch, L. A new principle of RNA folding based on pseudoknotting. Nucleic Acids Res. 1985, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Ucisik, M.N.; Bevilacqua, P.C.; Hammes-Schiffer, S. Molecular Dynamics Study of Twister Ribozyme: Role of Mg2+ Ions and the Hydrogen-Bonding Network in the Active Site. Biochemistry 2016, 55, 3834–3846. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Vusurovic, N.; Gebetsberger, J.; Gao, P.; Juen, M.; Kreutz, C.; Micura, R.; Patel, D.J. Pistol ribozyme adopts a pseudoknot fold facilitating site-specific in-line cleavage. Nat. Chem. Biol. 2016, 12, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.A.; Wang, J.; Steitz, T.A. Crystal structure of Pistol, a class of self-cleaving ribozyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Stahley, M.R.; Strobel, S.A. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 2005, 309, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Toor, N.; Keating, K.S.; Pyle, A.M. Structural insights into RNA splicing. Curr. Opin. Struct. Biol. 2009, 19, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, G.M.; Nakamura, S.; Roth, A.; Breaker, R.R. Ribozyme speed limits. RNA 2003, 9, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Raines, R.T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. [Google Scholar] [PubMed]
- Breaker, R.R.; Emilsson, G.M.; Lazarev, D.; Nakamura, S.; Puskarz, I.J.; Roth, A.; Sudarsan, N. A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA 2003, 9, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Pyle, A.M. Ribozymes: A distinct class of metalloenzymes. Science 1993, 261, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Gebetsberger, J.; Micura, R. Unwinding the twister ribozyme: From structure to mechanism. Wiley Interdiscip. Rev. RNA 2016. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Liu, Y.; Lilley, D.M. Ribozymes and the mechanisms that underlie RNA catalysis. Front. Chem. Sci. Eng. 2016, 10, 178–185. [Google Scholar] [CrossRef]
- Breaker, R.R. Mechanistic Debris Generated by Twister Ribozymes. ACS Chem. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rupert, P.B.; Massey, A.P.; Sigurdsson, S.T.; Ferre-D’Amare, A.R. Transition state stabilization by a catalytic RNA. Science 2002, 298, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.; Golden, B.L. Two Active Site Divalent Ions in the Crystal Structure of the Hammerhead Ribozyme Bound to a Transition State Analogue. Biochemistry 2016, 55, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Suslov, N.B.; DasGupta, S.; Huang, H.; Fuller, J.R.; Lilley, D.M.; Rice, P.A.; Piccirilli, J.A. Crystal structure of the Varkud satellite ribozyme. Nat. Chem. Biol. 2015, 11, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, J.C.; Lipchock, S.V.; Strobel, S.A. Structural investigation of the glmS ribozyme bound to Its catalytic cofactor. Chem. Biol. 2007, 14, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.; Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004, 429, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Das, S.R.; Piccirilli, J.A. General acid catalysis by the hepatitis delta virus ribozyme. Nat. Chem. Biol. 2005, 1, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Chadalavada, D.M.; Bevilacqua, P.C. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 2000, 287, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Silva Lopez, C.; Giambasu, G.M.; Martick, M.; Scott, W.G.; York, D.M. Role of Mg2+ in hammerhead ribozyme catalysis from molecular simulation. J. Am. Chem. Soc. 2008, 130, 3053–3064. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Perrin, D.M. Probing general acid catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 2009, 131, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Peebles, C.L.; Perlman, P.S.; Mecklenburg, K.L.; Petrillo, M.L.; Tabor, J.H.; Jarrell, K.A.; Cheng, H.L. A self-splicing RNA excises an intron lariat. Cell 1986, 44, 213–223. [Google Scholar] [CrossRef]
- Toor, N.; Keating, K.S.; Taylor, S.D.; Pyle, A.M. Crystal structure of a self-spliced group II intron. Science 2008, 320, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Marcia, M.; Pyle, A.M. Visualizing group II intron catalysis through the stages of splicing. Cell 2012, 151, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.V.; Krivenko, A.A.; Pace, N.R. Mapping metal-binding sites in the catalytic domain of bacterial RNase P RNA. RNA 2009, 15, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Yajima, R.; Chadalavada, D.M.; Chase, E.; Bevilacqua, P.C.; Golden, B.L. A 1.9 A crystal structure of the HDV ribozyme precleavage suggests both Lewis acid and general acid mechanisms contribute to phosphodiester cleavage. Biochemistry 2010, 49, 6508–6518. [Google Scholar] [CrossRef] [PubMed]
- Ferre-D’Amare, A.R.; Zhou, K.; Doudna, J.A. Crystal structure of a hepatitis delta virus ribozyme. Nature 1998, 395, 567–574. [Google Scholar] [PubMed]
- Mir, A.; Chen, J.; Robinson, K.; Lendy, E.; Goodman, J.; Neau, D.; Golden, B.L. Two Divalent Metal Ions and Conformational Changes Play Roles in the Hammerhead Ribozyme Cleavage Reaction. Biochemistry 2015, 54, 6369–6381. [Google Scholar] [CrossRef] [PubMed]
- Rupert, P.B.; Ferre-D’Amare, A.R. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature 2001, 410, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Fedor, M.J. Tertiary structure stabilization promotes hairpin ribozyme ligation. Biochemistry 1999, 38, 11040–11050. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.A. The Neurospora Varkud satellite ribozyme. Biochem. Soc. Trans. 2002, 30, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Moody, E.M.; Lecomte, J.T.; Bevilacqua, P.C. Linkage between proton binding and folding in RNA: A thermodynamic framework and its experimental application for investigating pKa shifting. RNA 2005, 11, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Pinard, R.; Hampel, K.J.; Heckman, J.E.; Lambert, D.; Chan, P.A.; Major, F.; Burke, J.M. Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J. 2001, 20, 6434–6442. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Ouellet, J.; Zhao, Z.Y.; Harusawa, S.; Araki, L.; Kurihara, T.; Lilley, D.M. Nucleobase catalysis in the hairpin ribozyme. RNA 2006, 12, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Kath-Schorr, S.; Wilson, T.J.; Li, N.S.; Lu, J.; Piccirilli, J.A.; Lilley, D.M. General acid-base catalysis mediated by nucleobases in the hairpin ribozyme. J. Am. Chem. Soc. 2012, 134, 16717–16724. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Li, N.S.; Lu, J.; Frederiksen, J.K.; Piccirilli, J.A.; Lilley, D.M. Nucleobase-mediated general acid-base catalysis in the Varkud satellite ribozyme. Proc. Natl. Acad. Sci. USA 2010, 107, 11751–11756. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; McLeod, A.C.; Lilley, D.M. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 2007, 26, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Liu, Y.; Domnick, C.; Kath-Schorr, S.; Lilley, D.M. The Novel Chemical Mechanism of the Twister Ribozyme. J. Am. Chem. Soc. 2016, 138, 6151–6162. [Google Scholar] [CrossRef] [PubMed]
- Kapinos, L.E.; Operschall, B.P.; Larsen, E.; Sigel, H. Understanding the acid-base properties of adenosine: The intrinsic basicities of N1, N3 and N7. Chemistry 2011, 17, 8156–8164. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Mehdizadeh, R.; Olive, J.E.; Collins, R.A. The ionic environment determines ribozyme cleavage rate by modulation of nucleobase pK a. RNA 2008, 14, 1942–1949. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.D.; Johnson, F.A.; Shafer, J.A. Effect of cysteine-25 on the ionization of histidine-159 in papain as determined by proton nuclear magnetic resonance spectroscopy. Evidence for a his-159–Cys-25 ion pair and its possible role in catalysis. Biochemistry 1981, 20, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Storer, A.C.; Menard, R. Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 1994, 244, 486–500. [Google Scholar] [PubMed]
- Draper, D.E. RNA folding: Thermodynamic and molecular descriptions of the roles of ions. Biophys. J. 2008, 95, 5489–5495. [Google Scholar] [CrossRef] [PubMed]
- Pontius, B.W.; Lott, W.B.; von Hippel, P.H. Observations on catalysis by hammerhead ribozymes are consistent with a two-divalent-metal-ion mechanism. Proc. Natl. Acad. Sci. USA 1997, 94, 2290–2294. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.A. Structural and catalytic chemistry of magnesium-dependent enzymes. Biometals 2002, 15, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.A. Metallobiochemistry of RNA. Co(NH3)63+ as a probe for Mg2+(aq) binding sites. J. Inorg. Biochem. 1993, 49, 171–175. [Google Scholar] [CrossRef]
- Hampel, A.; Cowan, J.A. A unique mechanism for RNA catalysis: The role of metal cofactors in hairpin ribozyme cleavage. Chem. Biol. 1997, 4, 513–517. [Google Scholar] [CrossRef]
- Roth, A.; Nahvi, A.; Lee, M.; Jona, I.; Breaker, R.R. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. RNA 2006, 12, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Sigel, R.K.; Pyle, A.M. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007, 107, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.G. Ribozyme catalysis revisited: Is water involved? Mol. Cell 2007, 28, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.K.; Cohn, M. Diastereomers of the nucleoside phosphorothioates as probes of the structure of the metal nucleotide substrates and of the nucleotide binding site of yeast hexokinase. J. Biol. Chem. 1979, 254, 10839–10845. [Google Scholar] [PubMed]
- Pecoraro, V.L.; Hermes, J.D.; Cleland, W.W. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry 1984, 23, 5262–5271. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ribozyme Family | Alignment of O2′ to P-O5′ Bond (α Catalysis) | Neutralization of the Negative Charge on a Non-Bridging Oxygen (β Catalysis) | Deprotonation of 2′ Nucleophile (γ Catalysis) | Protonation of 5′ Leaving Group (δ Catalysis) | Cleavage Rate (min−1) at Neutral pH | Ref. |
|---|---|---|---|---|---|---|
| Twister 1 (O. sativa) | 83° (4OJI) | G45 | G45 | A7 | 2.45 | [30] |
| Twister 1 (env9) | 83° (4QJH) | A63 | G62 | [28] | ||
| Twister 1 (env22) | 148° (4RGE) | G48, Mg2+ | G48? 2 | A6? 2 | 2.44 (at 20 °C) | [29,31] |
| 91° (5DUN) | Mg2+-bound water? 2 | 1.41 (at 15 °C) | ||||
| Pistol | 167° (5K7C) | G40 | G40 | A32 (or G32) | 2.72 (at 20 °C) | [34,35] |
| 126° (5KTJ) | 0.88 (at 15 °C) | |||||
| HDV | 140° (5DI2) | Mg2+ | Mg2+ | C75 | 52 | [60,61] |
| Hammerhead | G12 | G8, metal-bound water | 1.39 | [46,62] | ||
| Hairpin | G8 | G8 | A38 | 0.1–0.5 | [63,64] | |
| Neurospora VS | G638 | G638 | A756 | 1 | [23,65] | |
| glmS | GlcN6P | G40 | GlcN6P | 1–3 | [26,48] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-Y.; Lee, B.-J. Structural and Biochemical Properties of Novel Self-Cleaving Ribozymes. Molecules 2017, 22, 678. https://doi.org/10.3390/molecules22040678
Lee K-Y, Lee B-J. Structural and Biochemical Properties of Novel Self-Cleaving Ribozymes. Molecules. 2017; 22(4):678. https://doi.org/10.3390/molecules22040678
Chicago/Turabian StyleLee, Ki-Young, and Bong-Jin Lee. 2017. "Structural and Biochemical Properties of Novel Self-Cleaving Ribozymes" Molecules 22, no. 4: 678. https://doi.org/10.3390/molecules22040678
APA StyleLee, K. -Y., & Lee, B. -J. (2017). Structural and Biochemical Properties of Novel Self-Cleaving Ribozymes. Molecules, 22(4), 678. https://doi.org/10.3390/molecules22040678