
On the Many Actions of Ouabain: Pro-Cystogenic Effects in Autosomal Dominant Polycystic Kidney Disease

{kind=link}
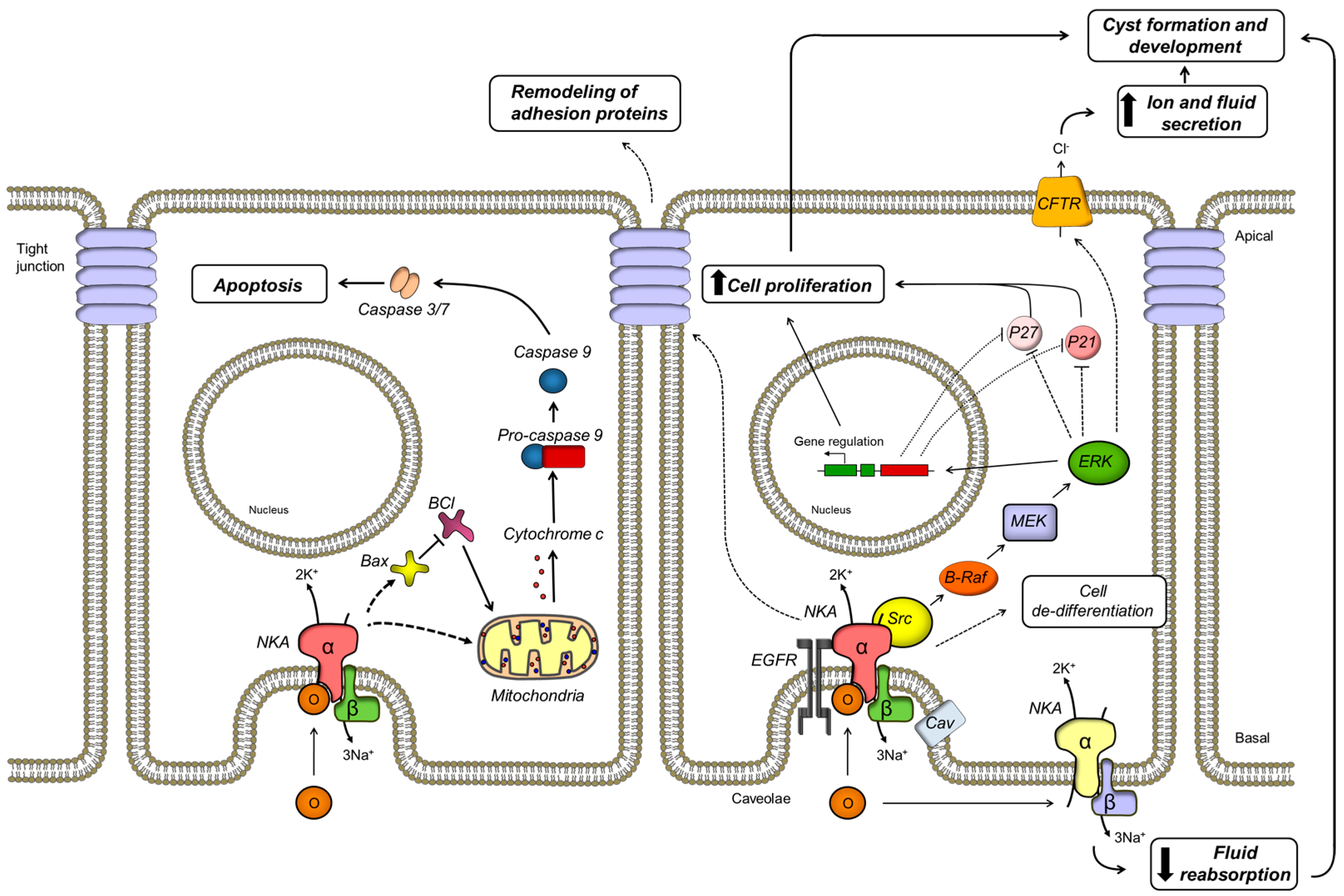
Abstract
:1. Ouabain Structure and Overall Activity
2. The Ouabain Target, Na,K-ATPase
3. Ouabain Actions in Different Cells and Tissues
4. Ouabain Activation of Cell Signaling
5. Ouabain as a Hormone
6. Ouabain and Autosomal Dominant Polycystic Kidney Disease
7. Pro-Cystogenic Actions of Ouabain in ADPKD
8. Ouabain and NKA Signaling as Targets for the Treatment of Disease
9. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Agrawal, A.A.; Petschenka, G.; Bingham, R.A.; Weber, M.G.; Rasmann, S. Toxic cardenolides: Chemical ecology and coevolution of specialized plant-herbivore interactions. New Phytol. 2012, 194, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Ziff, O.J.; Kotecha, D. Digoxin: The good and the bad. Trends Cardiovasc. Med. 2016, 26, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous cardiac glycosides: Hormones using the sodium pump as signal transducer. Semin. Nephrol. 2005, 25, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Buckalew, V.M. Endogenous digitalis-like factors. An historical overview. Front. Biosci. 2005, 10, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Diederich, M.; Muller, F.; Cerella, C. Cardiac glycosides: From molecular targets to immunogenic cell death. Biochem. Pharmacol. 2017, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nesher, M.; Shpolansky, U.; Rosen, H.; Lichtstein, D. The digitalis-like steroid hormones: New mechanisms of action and biological significance. Life Sci. 2007, 80, 2093–2107. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, R.I.; Doris, P.A. Cardiotonic steroids: Potential endogenous sodium pump ligands with diverse function. Exp. Biol. Med. 2002, 227, 561–569. [Google Scholar]
- Barrueto, F., Jr.; Kirrane, B.M.; Cotter, B.W.; Hoffman, R.S.; Nelson, L.S. Cardioactive steroid poisoning: A comparison of plant- and animal-derived compounds. J. Med. Toxicol. 2006, 2, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Goldman, P. Herbal medicines today and the roots of modern pharmacology. Ann. Intern. Med. 2001, 135, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Wachtel-Galor, S.; Benzie, I.F.F. Herbal Medicine: An Introduction to Its History, Usage, Regulation, Current Trends, and Research Needs. In Herbal Medicine: Biomolecular and Clinical Aspects; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Bessen, H.A. Therapeutic and toxic effects of digitalis: William Withering, 1785. J. Emerg. Med. 1986, 4, 243–2488. [Google Scholar] [CrossRef]
- Meijler, F.L. An “account” of digitalis and atrial fibrillation. J. Am. Coll. Cardiol. 1985, 5 (Suppl. S1), 60A–68A. [Google Scholar] [CrossRef]
- Katz, A.M. Effects of digitalis on cell biochemistry: Sodium pump inhibition. J. Am. Coll. Cardiol. 1985, 5 (Suppl. S1), 16A–21A. [Google Scholar] [CrossRef]
- Kaplan, J.H. Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 2002, 71, 511–535. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.V. William Withering revisited: 200 years of the foxglove. Am. J. Cardiol. 1986, 58, 189–190. [Google Scholar] [CrossRef]
- Smith, T.W. The basic mechanism of inotropic action of digitalis glycosides. J. Pharmacol. 1984, 15 (Suppl. S1), 35–51. [Google Scholar] [PubMed]
- McDonough, A.A.; Velotta, J.B.; Schwinger, R.H.; Philipson, K.D.; Farley, R.A. The cardiac sodium pump: Structure and function. Basic Res. Cardiol. 2002, 97 (Suppl. S1), I19–I24. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P. Sodium ions, calcium ions, blood pressure regulation, and hypertension: A reassessment and a hypothesis. Am. J. Physiol. 1977, 232, C165–C173. [Google Scholar] [PubMed]
- Blaustein, M.P.; Hamlyn, J.M. Signaling mechanisms that link salt retention to hypertension: Endogenous ouabain, the Na+ pump, the Na+/Ca2+ exchanger and TRPC proteins. Biochim. Biophys. Acta 2010, 1802, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Role of endogenous cardiotonic steroids in sodium homeostasis. Nephrol. Dial. Transplant. 2008, 23, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.L.; Skou, J.C. Preparation of highly active (Na+ + K+)-ATPase from the outer medulla of rabbit kidney. Biochem. Biophys. Res. Commun. 1969, 37, 39–46. [Google Scholar] [CrossRef]
- Fambrough, D.M. Studies on the Na+-K+ ATPase of skeletal muscle and nerve. Cold Spring Harb. Symp. Quant. Biol. 1983, 48, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Zahler, R.; Sun, W.; Ardito, T.; Zhang, Z.T.; Kocsis, J.D.; Kashgarian, M. The α3 isoform protein of the Na+, K+-ATPase is associated with the sites of cardiac and neuromuscular impulse transmission. Circ. Res. 1996, 78, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J. Isozymes of the Na+/K+-ATPase. Biochim. Biophys. Acta 1989, 988, 185–220. [Google Scholar] [CrossRef]
- Lingrel, J.B.; Kuntzweiler, T. Na+,K+-ATPase. J. Biol. Chem. 1994, 269, 19659–19662. [Google Scholar] [PubMed]
- Lindzen, M.; Gottschalk, K.E.; Fuzesi, M.; Garty, H.; Karlish, S.J. Structural interactions between FXYD proteins and Na+,K+-ATPase: α/β/FXYD subunit stoichiometry and cross-linking. J. Biol. Chem. 2006, 281, 5947–5955. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.L.; Hakansson, K.O.; Karlish, S.J. Structure and mechanism of Na,K-ATPase: Functional sites and their interactions. Annu. Rev. Physiol. 2003, 65, 817–849. [Google Scholar] [CrossRef] [PubMed]
- Apell, H.J.; Schneeberger, A.; Sokolov, V.S. Partial reactions of the Na,K-ATPase: Kinetic analysis and transport properties. Acta Physiol. Scand. Suppl. 1998, 643, 235–245. [Google Scholar] [PubMed]
- Ackermann, U.; Geering, K. Mutual dependence of Na,K-ATPase α- and β-subunits for correct posttranslational processing and intracellular transport. FEBS Lett. 1990, 269, 105–108. [Google Scholar] [CrossRef]
- Chow, D.C.; Forte, J.G. Functional significance of the β-subunit for heterodimeric P-type ATPases. J. Exp. Biol. 1995, 198, 1–17. [Google Scholar] [PubMed]
- Geering, K. Subunit assembly and functional maturation of Na,K-ATPase. J. Membr. Biol. 1990, 115, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.A.; Gopal, J.; Willis, D.; Espineda, C.; Twiss, J.L.; Rajasekaran, A.K. Na,K-ATPase β1-subunit increases the translation efficiency of the α-subunit in MSV-MDCK cells. Mol. Biol. Cell 2004, 15, 3224–3232. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Dada, L.A.; Tokhtaeva, E.; Sachs, G. The Na-K-ATPase α1β1 heterodimer as a cell adhesion molecule in epithelia. Am. J. Physiol. Cell Physiol. 2012, 302, C1271–C1281. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Larre, I. The Na+-K+-ATPase as self-adhesion molecule and hormone receptor. Am. J. Physiol. Cell Physiol. 2012, 302, C473–C481. [Google Scholar] [CrossRef] [PubMed]
- Muller-Husmann, G.; Gloor, S.; Schachner, M. Functional characterization of beta isoforms of murine Na,K-ATPase. The adhesion molecule on glia (AMOG/beta 2), but not beta 1, promotes neurite outgrowth. J. Biol. Chem. 1993, 268, 26260–26267. [Google Scholar] [PubMed]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial junctions depend on intercellular trans-interactions between the Na,K-ATPase β1 subunits. J. Biol. Chem. 2011, 286, 25801–25812. [Google Scholar] [CrossRef] [PubMed]
- Hardwicke, P.M.; Freytag, J.W. A proteolipid associated with Na,K-ATPase is not essential for ATPase activity. Biochem. Biophys. Res. Commun. 1981, 102, 250–257. [Google Scholar] [CrossRef]
- Garty, H.; Karlish, S.J. Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 2006, 68, 431–459. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. FXYD proteins: New regulators of Na-K-ATPase. Am. J. Physiol. Ren. Physiol. 2006, 290, F241–F250. [Google Scholar] [CrossRef] [PubMed]
- Schmalzing, G.; Ruhl, K.; Gloor, S.M. Isoform-specific interactions of Na,K-ATPase subunits are mediated via extracellular domains and carbohydrates. Proc. Natl. Acad. Sci. USA 1997, 94, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Avila, J.; Cozar-Castellano, I.; Brownleader, M.D.; Trevan, M.; Francis, M.J.; Lamb, J.F.; Martin-Vasallo, P. Na+, K+-ATPase isozyme diversity; comparative biochemistry and physiological implications of novel functional interactions. Biosci. Rep. 2000, 20, 51–91. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J. Overview: Subunit diversity in the Na,K-ATPase. Soc. Gen. Physiol. Ser. 1991, 46, 63–76. [Google Scholar] [PubMed]
- Keryanov, S.; Gardner, K.L. Physical mapping and characterization of the human Na,K-ATPase isoform, ATP1A4. Gene 2002, 292, 151–166. [Google Scholar] [CrossRef]
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 2005, 25, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Pu, H.X.; Cluzeaud, F.; Goldshleger, R.; Karlish, S.J.; Farman, N.; Blostein, R. Functional role and immunocytochemical localization of the γa and γb forms of the Na,K-ATPase gamma subunit. J. Biol. Chem. 2001, 276, 20370–20378. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Levy-Holzman, R.; Cluzeaud, F.; Farman, N.; Garty, H. Membrane topology and immunolocalization of CHIF in kidney and intestine. Am. J. Physiol. Ren. Physiol. 2001, 280, F505–F512. [Google Scholar]
- Sweadner, K.J.; Rael, E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 2000, 68, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Eakle, K.A.; Kabalin, M.A.; Wang, S.G.; Farley, R.A. The influence of beta subunit structure on the stability of Na+/K+-ATPase complexes and interaction with K+. J. Biol. Chem. 1994, 269, 6550–6557. [Google Scholar] [PubMed]
- Blanco, G.; Koster, J.C.; Sanchez, G.; Mercer, R.W. Kinetic properties of the alpha2.beta.1 and alpha.2.beta.2 isozymes of the Na,K-ATPase. Biochemistry 1995, 34, 319–325. [Google Scholar] [PubMed]
- Blanco, G.; Sanchez, G.; Mercer, R.W. Comparison of the enzymatic properties of the Na,K-ATPase alpha 3 beta 1 and alpha 3 beta 2 isozymes. Biochemistry 1995, 34, 9897–9903. [Google Scholar] [CrossRef] [PubMed]
- Crambert, G.; Hasler, U.; Beggah, A.T.; Yu, C.; Modyanov, N.N.; Horisberger, J.D.; Lelievre, L.; Geering, K. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J. Biol. Chem. 2000, 275, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650. [Google Scholar] [PubMed]
- Riganti, C.; Campia, I.; Kopecka, J.; Gazzano, E.; Doublier, S.; Aldieri, E.; Bosia, A.; Ghigo, D. Pleiotropic effects of cardioactive glycosides. Curr. Med. Chem. 2011, 18, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Silva, E.; Soares-da-Silva, P. New insights into the regulation of Na+,K+-ATPase by ouabain. Int. Rev. Cell Mol. Biol. 2012, 294, 99–132. [Google Scholar] [PubMed]
- Kometiani, P.; Li, J.; Gnudi, L.; Kahn, B.B.; Askari, A.; Xie, Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 15249–15256. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Lopatina, E.V.; Kipenko, A.V.; Pasatetskaya, N.A.; Penniyaynen, V.A.; Krylov, B.V. Modulation of the transducer function of Na+,K+-ATPase: New mechanism of heart remodeling. Can. J. Physiol. Pharmacol. 2016, 94, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Belliard, A.; Gulati, G.K.; Duan, Q.; Alves, R.; Brewer, S.; Madan, N.; Sottejeau, Y.; Wang, X.; Kalisz, J.; Pierre, S.V. Ischemia/reperfusion-induced alterations of enzymatic and signaling functions of the rat cardiac Na+/K+-ATPase: Protection by ouabain preconditioning. Physiol. Rep. 2016, 4, e12991. [Google Scholar] [CrossRef] [PubMed]
- Belliard, A.; Sottejeau, Y.; Duan, Q.; Karabin, J.L.; Pierre, S.V. Modulation of cardiac Na+,K+-ATPase cell surface abundance by simulated ischemia-reperfusion and ouabain preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H94–H103. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.C.; Abramowitz, J.; Koksoy, A. Low concentrations of ouabain activate vascular smooth muscle cell proliferation. Ann. N. Y. Acad. Sci. 2003, 986, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Q.; Guan, L. Effects of ouabain on proliferation, intracellular free calcium and c-myc mRNA expression in vascular smooth muscle cells. J. Comp. Physiol. B 2007, 177, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, J.; Dai, C.; Hirschi, K.K.; Dmitrieva, R.I.; Doris, P.A.; Liu, L.; Allen, J.C. Ouabain- and marinobufagenin-induced proliferation of human umbilical vein smooth muscle cells and a rat vascular smooth muscle cell line, A7r5. Circulation 2003, 108, 3048–3053. [Google Scholar] [CrossRef] [PubMed]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Hangaard, L.; Bouzinova, E.V.; Staehr, C.; Dam, V.S.; Kim, S.; Xie, Z.; Aalkjaer, C.; Matchkov, V.V. Na,K-ATPase regulates intercellular communication in the vascular wall via cSrc kinase dependent connexin43 phosphorylation. Am. J. Physiol. Cell Physiol. 2017, 312, C385–C397. [Google Scholar] [CrossRef] [PubMed]
- Khundmiri, S.J.; Metzler, M.A.; Ameen, M.; Amin, V.; Rane, M.J.; Delamere, N.A. Ouabain induces cell proliferation through calcium-dependent phosphorylation of Akt (protein kinase B) in opossum kidney proximal tubule cells. Am. J. Physiol. Cell Physiol. 2006, 291, C1247–C1257. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, R.I.; Doris, P.A. Ouabain is a potent promoter of growth and activator of ERK1/2 in ouabain-resistant rat renal epithelial cells. J. Biol. Chem. 2003, 278, 28160–28166. [Google Scholar] [PubMed]
- Montero, A.; Rodriguez-Barbero, A.; Ricote, M.; Sancho, J.; Lopez-Novoa, J.M. Effect of ouabain and hypothalamic, hypophysary inhibitory factor on rat mesangial cell proliferation. J. Cardiovasc. Pharmacol. 1993, 22 (Suppl. S2), S35–S37. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wu, L.; Qu, W.; Malhotra, D.; Xie, Z.; Shapiro, J.I.; Liu, J. Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 2008, 294, C555–C563. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.G.; Flores-Maldonado, C.; Lazaro, A.; Shoshani, L.; Flores-Benitez, D.; Larre, I.; Cereijido, M. Ouabain binding to Na+,K+-ATPase relaxes cell attachment and sends a specific signal (NACos) to the nucleus. J. Membr. Biol. 2004, 198, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Larre, I.; Lazaro, A.; Contreras, R.G.; Balda, M.S.; Matter, K.; Flores-Maldonado, C.; Ponce, A.; Flores-Benitez, D.; Rincon-Heredia, R.; Padilla-Benavides, T.; et al. Ouabain modulates epithelial cell tight junction. Proc. Natl. Acad. Sci. USA 2010, 107, 11387–11392. [Google Scholar] [CrossRef] [PubMed]
- Larre, I.; Castillo, A.; Flores-Maldonado, C.; Contreras, R.G.; Galvan, I.; Munoz-Estrada, J.; Cereijido, M. Ouabain modulates ciliogenesis in epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20591–20596. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Payne, K.; Lee-Kwon, W.; Zhang, Z.; Lim, S.W.; Hamlyn, J.; Blaustein, M.P.; Kwon, H.M.; Pallone, T.L. Chronic ouabain treatment induces vasa recta endothelial dysfunction in the rat. Am. J. Physiol. Ren. Physiol. 2009, 296, F98–F106. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Khodus, G.R.; Kruusmagi, M.; Kamali-Zare, P.; Liu, X.L.; Eklof, A.C.; Zelenin, S.; Brismar, H.; Aperia, A. Ouabain protects against adverse developmental programming of the kidney. Nat. Commun. 2010, 1, 42. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zelenin, S.; Aperia, A.; Aizman, O. Low doses of ouabain protect from serum deprivation-triggered apoptosis and stimulate kidney cell proliferation via activation of NF-κB. J. Am. Soc. Nephrol. 2006, 17, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Salles von-Held-Ventura, J.; Mazala-de-Oliveira, T.; Candida da Rocha Oliveira, A.; Granja, M.G.; Goncalves-de-Albuquerque, C.F.; Castro-Faria-Neto, H.C.; Giestal-de-Araujo, E. The trophic effect of ouabain on retinal ganglion cells is mediated by IL-1β and TNF-α. Biochem. Biophys. Res. Commun. 2016, 478, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, G.M. Interneuron regeneration after ouabain treatment in the adult mammalian retina. Neuroreport 2015, 26, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Lopachev, A.V.; Lopacheva, O.M.; Osipova, E.A.; Vladychenskaya, E.A.; Smolyaninova, L.V.; Fedorova, T.N.; Koroleva, O.V.; Akkuratov, E.E. Ouabain-induced changes in MAP kinase phosphorylation in primary culture of rat cerebellar cells. Cell Biochem. Funct. 2016, 34, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Dvela, M.; Rosen, H.; Ben-Ami, H.C.; Lichtstein, D. Endogenous ouabain regulates cell viability. Am. J. Physiol. Cell Physiol. 2012, 302, C442–C452. [Google Scholar] [CrossRef] [PubMed]
- Winnicka, K.; Bielawski, K.; Bielawska, A.; Miltyk, W. Dual effects of ouabain, digoxin and proscillaridin A on the regulation of apoptosis in human fibroblasts. Nat. Prod. Res. 2010, 24, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.F.; Amaral, L.S.; Porto, C.S.; Quintas, L.E. Na+/K+-ATPase alpha1 isoform mediates ouabain-induced expression of cyclin D1 and proliferation of rat sertoli cells. Reproduction 2012, 144, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Xiong, A.X.; Wang, M.; Jin, R.M.; Bai, Y.; Lin, W. Ouabain-induced apoptosis of Jurkat cells correlates with activation of caspase-3 and regulation of Bcl-2 gene family. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006, 14, 891–894. [Google Scholar] [PubMed]
- Esteves, M.B.; Marques-Santos, L.F.; Affonso-Mitidieri, O.R.; Rumjanek, V.M. Ouabain exacerbates activation-induced cell death in human peripheral blood lymphocytes. An. Acad. Bras. Cienc. 2005, 77, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Olej, B.; dos Santos, N.F.; Leal, L.; Rumjanek, V.M. Ouabain induces apoptosis on PHA-activated lymphocytes. Biosci. Rep. 1998, 18, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brodie, C.; Tordai, A.; Saloga, J.; Domenico, J.; Gelfand, E.W. Ouabain induces inhibition of the progression phase in human T-cell proliferation. J. Cell. Physiol. 1995, 165, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, C.R.; Valente, R.C.; Echevarria-Lima, J.; Fontes, C.F.; de Araujo-Martins, L.; Araujo, E.G.; Rumjanek, V.M. The influence of Ouabain on human dendritic cells maturation. Mediat. Inflamm. 2014, 2014, 494956. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.P.; Rumjanek, V.M. Ouabain affects the expression of activation markers, cytokine production, and endocytosis of human monocytes. Mediat. Inflamm. 2014, 2014, 760368. [Google Scholar] [CrossRef] [PubMed]
- Kotova, O.; Galuska, D.; Essen-Gustavsson, B.; Chibalin, A.V. Metabolic and signaling events mediated by cardiotonic steroid ouabain in rat skeletal muscle. Cell. Mol. Biol. 2006, 52, 48–57. [Google Scholar] [PubMed]
- Al-Ghoul, M.; Valdes, R., Jr. Mammalian cardenolides in cancer prevention and therapeutics. Ther. Drug Monit. 2008, 30, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.A.; Yang, P.; Pawlus, A.D.; Block, K.I. Cardiac glycosides as novel cancer therapeutic agents. Mol. Interv. 2008, 8, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Na+/K+-ATPase and cancer. Pharm. Pat. Anal. 2012, 1, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 2007, 1776, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Montano, J.M.; Burgos-Moron, E.; Orta, M.L.; Maldonado-Navas, D.; Garcia-Dominguez, I.; Lopez-Lazaro, M. Evaluating the cancer therapeutic potential of cardiac glycosides. Biomed. Res. Int. 2014, 2014, 794930. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Cardiotonic steroids-mediated targeting of the Na+/K+-ATPase to combat chemoresistant cancers. Curr. Med. Chem. 2012, 19, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Cerella, C.; Guchelaar, H.J.; Diederich, M.; Gelderblom, H. Cardiac glycosides in cancer therapy: From preclinical investigations towards clinical trials. Investig. New Drugs 2013, 31, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Winnicka, K.; Bielawski, K.; Bielawska, A. Cardiac glycosides in cancer research and cancer therapy. Acta Pol. Pharm. 2006, 63, 109–115. [Google Scholar] [PubMed]
- Winnicka, K.; Bielawski, K.; Bielawska, A.; Surazynski, A. Antiproliferative activity of derivatives of ouabain, digoxin and proscillaridin A in human MCF-7 and MDA-MB-231 breast cancer cells. Biol. Pharm. Bull. 2008, 31, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.A.; Silva, K.A.; Delou, J.M.; Fonseca, L.M.; Lopes, A.G.; Capella, M.A. Modulation of ABCC1 and ABCG2 proteins by ouabain in human breast cancer cells. Anticancer Res. 2014, 34, 1441–1448. [Google Scholar]
- Magpusao, A.N.; Omolloh, G.; Johnson, J.; Gascon, J.; Peczuh, M.W.; Fenteany, G. Cardiac glycoside activities link Na+/K+ ATPase ion-transport to breast cancer cell migration via correlative SAR. ACS Chem. Biol. 2015, 10, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Ninsontia, C.; Chanvorachote, P. Ouabain mediates integrin switch in human lung cancer cells. Anticancer Res. 2014, 34, 5495–5502. [Google Scholar] [PubMed]
- Ruanghirun, T.; Pongrakhananon, V.; Chanvorachote, P. Ouabain enhances lung cancer cell detachment. Anticancer Res. 2014, 34, 2231–2238. [Google Scholar] [PubMed]
- Trenti, A.; Grumati, P.; Cusinato, F.; Orso, G.; Bonaldo, P.; Trevisi, L. Cardiac glycoside ouabain induces autophagic cell death in non-small cell lung cancer cells via a JNK-dependent decrease of Bcl-2. Biochem. Pharmacol. 2014, 89, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Pongrakhananon, V.; Chunhacha, P.; Chanvorachote, P. Ouabain suppresses the migratory behavior of lung cancer cells. PLoS ONE 2013, 8, e68623. [Google Scholar] [CrossRef] [PubMed]
- Gasper, R.; Vandenbussche, G.; Goormaghtigh, E. Ouabain-induced modifications of prostate cancer cell lipidome investigated with mass spectrometry and FTIR spectroscopy. Biochim. Biophys. Acta 2011, 1808, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.D.; Mawji, I.A.; Anyiwe, K.; Williams, M.A.; Wang, X.; Venugopal, A.L.; Gronda, M.; Hurren, R.; Cheng, S.; Serra, S.; et al. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Cancer Res. 2009, 69, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Cuozzo, F.; Raciti, M.; Bertelli, L.; Parente, R.; Di Renzo, L. Pro-death and pro-survival properties of ouabain in U937 lymphoma derived cells. J. Exp. Clin. Cancer Res. 2012, 31, 95. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Wen, Y.; Zhou, M.; Li, J.; Wang, T.; Xu, P.; Ouyang, J. Ouabain induces apoptosis and autophagy in Burkitt’s lymphoma Raji cells. Biomed. Pharmacother. 2016, 84, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.W.; Jin, R.M.; Li, E.Q.; Wang, Y.R.; Bai, Y. Signal pathways in ouabain-induced proliferation of leukemia cells. World J. Pediatr. 2009, 5, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Wolle, D.; Lee, S.J.; Li, Z.; Litan, A.; Barwe, S.P.; Langhans, S.A. Inhibition of epidermal growth factor signaling by the cardiac glycoside ouabain in medulloblastoma. Cancer Med. 2014, 3, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liang, F.; Li, D.; Zheng, J. Ouabain elicits human glioblastoma cells apoptosis by generating reactive oxygen species in ERK-p66SHC-dependent pathway. Mol. Cell. Biochem. 2015, 398, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Kulikov, A.; Eva, A.; Kirch, U.; Boldyrev, A.; Scheiner-Bobis, G. Ouabain activates signaling pathways associated with cell death in human neuroblastoma. Biochim. Biophys. Acta 2007, 1768, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- De Souza, W.F.; Barbosa, L.A.; Liu, L.; de Araujo, W.M.; de-Freitas-Junior, J.C.; Fortunato-Miranda, N.; Fontes, C.F.; Morgado-Diaz, J.A. Ouabain-induced alterations of the apical junctional complex involve alpha1 and beta1 Na,K-ATPase downregulation and ERK1/2 activation independent of caveolae in colorectal cancer cells. J. Membr. Biol. 2014, 247, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Pezzani, R.; Rubin, B.; Redaelli, M.; Radu, C.; Barollo, S.; Cicala, M.V.; Salva, M.; Mian, C.; Mucignat-Caretta, C.; Simioni, P.; et al. The antiproliferative effects of ouabain and everolimus on adrenocortical tumor cells. Endocr. J. 2014, 61, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, F.; Fan, F.; Gu, Y.; Shan, N.; Meng, X.; Cheng, S.; Liu, Y.; Wang, C.; Song, Y.; et al. Quantitative Proteomics Reveals That the Inhibition of Na+/K+-ATPase Activity Affects S-Phase Progression Leading to a Chromosome Segregation Disorder by Attenuating the Aurora A Function in Hepatocellular Carcinoma Cells. J. Proteome Res. 2015, 14, 4594–4602. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Rhea, P.; Tan, L.; Cartwright, C.; Lee, H.J.; Ravoori, M.K.; Addington, C.; Gagea, M.; Kundra, V.; Kim, S.J.; et al. PBI-05204, a supercritical CO2 extract of Nerium oleander, inhibits growth of human pancreatic cancer via targeting the PI3K/mTOR pathway. Investig. New Drugs 2015, 33, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Kiss, R. The sodium pump alpha1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia 2008, 10, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Northfelt, D.W.; Chalasani, P.; Rensvold, D.; Kurtin, S.; Von Hoff, D.D.; Borad, M.J.; Tibes, R. Phase I trial of UNBS5162, a novel naphthalimide in patients with advanced solid tumors or lymphoma. Int. J. Clin. Oncol. 2013, 18, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G.; Wallace, D.P. Novel role of ouabain as a cystogenic factor in autosomal dominant polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 305, F797–F812. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Xie, Z. The Na,K-ATPase receptor complex: Its organization and membership. Cell Biochem. Biophys. 2006, 46, 303–316. [Google Scholar] [CrossRef]
- Ye, Q.; Li, Z.; Tian, J.; Xie, J.X.; Liu, L.; Xie, Z. Identification of a potential receptor that couples ion transport to protein kinase activity. J. Biol. Chem. 2011, 286, 6225–6232. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na+/K+-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Contreras, R.G.; Shoshani, L.; Flores-Maldonado, C.; Lazaro, A.; Cereijido, M. Relationship between Na+,K+-ATPase and cell attachment. J. Cell Sci. 1999, 112, 4223–4232. [Google Scholar] [PubMed]
- Rincon-Heredia, R.; Flores-Benitez, D.; Flores-Maldonado, C.; Bonilla-Delgado, J.; Garcia-Hernandez, V.; Verdejo-Torres, O.; Castillo, A.M.; Larre, I.; Poot-Hernandez, A.C.; Franco, M. Ouabain induces endocytosis and degradation of tight junction proteins through ERK1/2-dependent pathways. Exp. Cell Res. 2014, 320, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, K.; Kometiani, P.; Xie, Z.; Askari, A. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J. Biol. Chem. 2001, 276, 42050–42056. [Google Scholar] [CrossRef] [PubMed]
- Khundmiri, S.J.; Amin, V.; Henson, J.; Lewis, J.; Ameen, M.; Rane, M.J.; Delamere, N.A. Ouabain stimulates protein kinase B (Akt) phosphorylation in opossum kidney proximal tubule cells through an ERK-dependent pathway. Am. J. Physiol. Cell Physiol. 2007, 293, C1171–C1180. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Sottejeau, Y.; Gourbeau, J.M.; Sanchez, G.; Shidyak, A.; Blanco, G. Isoform specificity of Na-K-ATPase-mediated ouabain signaling. Am. J. Physiol. Ren. Physiol. 2008, 294, F859–F866. [Google Scholar] [CrossRef] [PubMed]
- Quintas, L.E.; Pierre, S.V.; Liu, L.; Bai, Y.; Liu, X.; Xie, Z.J. Alterations of Na+/K+-ATPase function in caveolin-1 knockout cardiac fibroblasts. J. Mol. Cell. Cardiol. 2010, 49, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Morgan, E.; Giovannucci, D.R.; Pierre, S.V.; Philipson, K.D.; Liu, L. Different roles of the cardiac Na+/Ca2+-exchanger in ouabain-induced inotropy, cell signaling, and hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H427–H435. [Google Scholar]
- Wu, J.; Akkuratov, E.E.; Bai, Y.; Gaskill, C.M.; Askari, A.; Liu, L. Cell signaling associated with Na+/K+-ATPase: Activation of phosphatidylinositide 3-kinase IA/Akt by ouabain is independent of Src. Biochemistry 2013, 52, 9059–9067. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009, 457, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ye, Q.; Cui, X.; Madan, N.; Yi, Q.; Pierre, S.V.; Xie, Z. Expression of rat Na-K-ATPase α2 enables ion pumping but not ouabain-induced signaling in α1-deficient porcine renal epithelial cells. Am. J. Physiol. Cell Physiol. 2015, 309, C373–C382. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P.; Zhang, J.; Chen, L.; Hamilton, B.P. How does salt retention raise blood pressure? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R514–R523. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Manunta, P. Ouabain-like factor: Is this the natriuretic hormone? Curr. Opin. Nephrol. Hypertens. 2000, 9, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, E.M.; Talner, L.B.; De Wardener, H.E. The effect of plasma from blood volume expanded dogs on sodium and potassium transport of renal tubule fragments. Clin. Sci. 1970, 38, 34P. [Google Scholar] [CrossRef] [PubMed]
- Tymiak, A.A.; Norman, J.A.; Bolgar, M.; DiDonato, G.C.; Lee, H.; Parker, W.L.; Lo, L.C.; Berova, N.; Nakanishi, K.; Haber, E.; et al. Physicochemical characterization of a ouabain isomer isolated from bovine hypothalamus. Proc. Natl. Acad. Sci. USA 1993, 90, 8189–8193. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P.; Bova, S.; DuCharme, D.W.; Harris, D.W.; Mandel, F.; Mathews, W.R.; Ludens, J.H. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. USA 1991, 88, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Bova, S.; Blaustein, M.P.; Ludens, J.H.; Harris, D.W.; DuCharme, D.W.; Hamlyn, J.M. Effects of an endogenous ouabainlike compound on heart and aorta. Hypertension 1991, 17, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nishimura, N.; Munakata, M.; Mori, T.; Okuda, K.; Nishino, N.; Hirose, S.; Kosaka, C.; Masuda, M.; Takahashi, H. Identification of endogenous ouabain in culture supernatant of PC12 cells. J. Hypertens. 2001, 19, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Laredo, J.; Hamilton, B.P.; Hamlyn, J.M. Secretion of endogenous ouabain from bovine adrenocortical cells: Role of the zona glomerulosa and zona fasciculata. Biochem. Biophys. Res. Commun. 1995, 212, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Haupert, G.T., Jr. Regulation of Na+, K+-ATPase by the endogenous sodium transport inhibitor from hypothalamus. Hypertension 1987, 10, I61–I66. [Google Scholar] [CrossRef] [PubMed]
- Budzikowski, A.S.; Huang, B.S.; Leenen, F.H. Brain “ouabain”, a neurosteroid, mediates sympathetic hyperactivity in salt-sensitive hypertension. Clin. Exp. Hypertens. 1998, 20, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Nesher, M.; Dvela, M.; Igbokwe, V.U.; Rosen, H.; Lichtstein, D. Physiological roles of endogenous ouabain in normal rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2026–H2034. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Lakatta, E.G.; Bagrov, A.Y. Endogenous Na,K pump ligands are differentially regulated during acute NaCl loading of Dahl rats. Circulation 2000, 102, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Structure-activity relationships for the hypertensinogenic activity of ouabain: Role of the sugar and lactone ring. Hypertension 2001, 37, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Lichtstein, D.; Steinitz, M.; Gati, I.; Samuelov, S.; Deutsch, J.; Orly, J. Biosynthesis of digitalis-like compounds in rat adrenal cells: Hydroxycholesterol as possible precursor. Life Sci. 1998, 62, 2109–2126. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Leenen, F.H.; Chen, L.; Golovina, V.A.; Hamlyn, J.M.; Pallone, T.L.; Van Huysse, J.W.; Zhang, J.; Wier, W.G. How NaCl raises blood pressure: A new paradigm for the pathogenesis of salt-dependent hypertension. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1031–H1049. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Yandle, T.G.; Hilton, P.J.; Jensen, B.P.; Begg, E.J.; Nicholls, M.G. Endogenous ouabain is not ouabain. Hypertension 2014, 64, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Fuerstenwerth, H. On the differences between ouabain and digitalis glycosides. Am. J. Ther. 2014, 21, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Ferrandi, M.; Bianchi, G.; Hamlyn, J.M. Endogenous ouabain in cardiovascular function and disease. J. Hypertens. 2009, 27, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Messaggio, E.; Casamassima, N.; Gatti, G.; Carpini, S.D.; Zagato, L.; Hamlyn, J.M. Endogenous ouabain in renal Na+ handling and related diseases. Biochim. Biophys. Acta 2010, 1802, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P. Salt sensitivity, endogenous ouabain and hypertension. Curr. Opin. Nephrol. Hypertens. 2013, 22, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Delva, P.; Capra, C.; Degan, M.; Minuz, P.; Covi, G.; Milan, L.; Steele, A.; Lechi, A. High plasma levels of a ouabain-like factor in normal pregnancy and in pre-eclampsia. Eur. J. Clin. Investig. 1989, 19, 95–100. [Google Scholar]
- Hasegawa, T.; Masugi, F.; Ogihara, T.; Kumahara, Y. Increase in plasma ouabainlike inhibitor of Na+, K+-ATPase with high sodium intake in patients with essential hypertension. J. Clin. Hypertens. 1987, 3, 419–429. [Google Scholar] [PubMed]
- Manunta, P.; Hamilton, B.P.; Hamlyn, J.M. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R553–R559. [Google Scholar] [CrossRef] [PubMed]
- Gabor, A.; Leenen, F.H. Mechanisms mediating sodium-induced pressor responses in the PVN of Dahl rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1338–R1349. [Google Scholar] [CrossRef] [PubMed]
- Loreaux, E.L.; Kaul, B.; Lorenz, J.N.; Lingrel, J.B. Ouabain-Sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J. Am. Soc. Nephrol. 2008, 19, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Haller, S.; Shapiro, A.; Malhotra, N.; Tian, J.; Xie, Z.; Malhotra, D.; Shapiro, J.I.; Liu, J. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Mol. Cell. Biochem. 2012, 367, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, R.; Ferrario, R.G.; Salvati, P.; Bianchi, G. Differences in ouabain-induced natriuresis between isolated kidneys of Milan hypertensive and normotensive rats. Clin. Sci. 1992, 82, 185–190. [Google Scholar] [CrossRef] [PubMed]
- McDougall, J.G.; Yates, N.A. Natriuresis and inhibition of Na+/K+-ATPase: Modulation of response by physiological manipulation. Clin. Exp. Pharmacol. Physiol. Suppl. 1998, 25, S57–S60. [Google Scholar] [CrossRef] [PubMed]
- Jaitovich, A.; Bertorello, A.M. Salt, Na+,K+-ATPase and hypertension. Life Sci. 2010, 86, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Tentori, S.; Ferrario, R.G.; Salvati, P.; Bianchi, G. Endogenous ouabain and aldosterone are coelevated in the circulation of patients with essential hypertension. J. Hypertens. 2016, 34, 2074–2080. [Google Scholar] [CrossRef] [PubMed]
- Hauck, C.; Frishman, W.H. Systemic hypertension: The roles of salt, vascular Na+/K+ ATPase and the endogenous glycosides, ouabain and marinobufagenin. Cardiol. Rev. 2012, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Skoumal, R.; Szokodi, I.; Aro, J.; Foldes, G.; Gooz, M.; Seres, L.; Sarman, B.; Lako-Futo, Z.; Papp, L.; Vuolteenaho, O.; et al. Involvement of endogenous ouabain-like compound in the cardiac hypertrophic process in vivo. Life Sci. 2007, 80, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.H.; Gao, H.Q.; Ji, X.; Wang, Y.; Liu, X.J.; You, B.A.; Cui, X.P.; Qiu, J. Effect of ouabain on myocardial ultrastructure and cytoskeleton during the development of ventricular hypertrophy. Heart Vessels 2013, 28, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wansapura, A.N.; Lasko, V.M.; Lingrel, J.B.; Lorenz, J.N. Mice expressing ouabain-sensitive β1-Na,K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H347–H355. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.; Manunta, P.; Casamassima, N.; Messaggio, E.; Jin, Y.; Thijs, L.; Richart, T.; Fagard, R.H.; Bianchi, G.; Staessen, J.A. Left ventricular geometry and endogenous ouabain in a Flemish population. J. Hypertens. 2009, 27, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Neglia, D.; Ghione, S.; D’Urso, G.; Baldacchino, M.C.; Montali, U.; L’Abbate, A. Increased circulating levels of ouabain-like factor in patients with asymptomatic left ventricular dysfunction. Eur. J. Heart Fail. 2001, 3, 165–171. [Google Scholar] [CrossRef]
- Pitzalis, M.V.; Hamlyn, J.M.; Messaggio, E.; Iacoviello, M.; Forleo, C.; Romito, R.; de Tommasi, E.; Rizzon, P.; Bianchi, G.; Manunta, P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur. J. Heart Fail. 2006, 8, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, D.; Du, L.; Baldawi, M.; Gable, M.E.; Askari, A.; Liu, L. Ouabain prevents pathological cardiac hypertrophy and heart failure through activation of phosphoinositide 3-kinase alpha in mouse. Cell Biosci. 2015, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Li, D.; Du, L.; Baldawi, M.; Gable, M.E.; Askari, A.; Liu, L. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: Relationship with plasma renin, aldosterone and blood pressure levels. J. Hypertens. 1995, 13, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Dostanic-Larson, I.; Neumann, J.C.; Lingrel, J.B. The ouabain-binding site of the α2 isoform of Na,K-ATPase plays a role in blood pressure regulation during pregnancy. Am. J. Hypertens. 2010, 23, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Villa, L.; Buono, R.; Ferrandi, M.; Molinari, I.; Benigni, F.; Bettiga, A.; Colciago, G.; Ikehata, M.; Messaggio, E.; Rastaldi, M.P. Ouabain Contributes to Kidney Damage in a Rat Model of Renal Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2016, 17, 1728. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.; Mullen, A.M.; McMahon, A.C.; Dawnay, A. Plasma OLC is elevated in mild experimental uremia but is not associated with hypertension. Am. J. Hypertens. 2001, 14, 1112–1115. [Google Scholar] [CrossRef]
- Stella, P.; Manunta, P.; Mallamaci, F.; Melandri, M.; Spotti, D.; Tripepi, G.; Hamlyn, J.M.; Malatino, L.S.; Bianchi, G.; Zoccali, C. Endogenous ouabain and cardiomyopathy in dialysis patients. J. Intern. Med. 2008, 263, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Manunta, P. Endogenous cardiotonic steroids in kidney failure: A review and an hypothesis. Adv. Chronic Kidney Dis. 2015, 22, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.N.; Wallace, D.P.; Blanco, G. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J. Am. Soc. Nephrol. 2007, 18, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Audrezet, M.P.; Le Meur, Y.; Chen, J.M.; Ferec, C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum. Mutat. 2014, 35, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, C.G.; Torres, V.E.; Offord, K.P.; Holley, K.E.; Beard, C.M.; Kurland, L.T. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney Dis. 1983, 2, 630–639. [Google Scholar] [CrossRef]
- Dalgaard, O.Z. Bilateral polycystic disease of the kidneys; a follow-up of two hundred and eighty-four patients and their families. Acta Med. Scand. Suppl. 1957, 328, 1–255. [Google Scholar] [PubMed]
- Grantham, J.J.; Cook, L.T.; Wetzel, L.H.; Cadnapaphornchai, M.A.; Bae, K.T. Evidence of extraordinary growth in the progressive enlargement of renal cysts. Clin. J. Am. Soc. Nephrol. 2010, 5, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Shokeir, M.H. Expression of “adult” polycystic renal disease in the fetus and newborn. Clin. Genet. 1978, 14, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Meijer, E.; Rook, M.; Tent, H.; Navis, G.; van der Jagt, E.J.; de Jong, P.E.; Gansevoort, R.T. Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J. Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int. Suppl. 1997, 63, S93–S97. [Google Scholar] [PubMed]
- Choukroun, G.; Itakura, Y.; Albouze, G.; Christophe, J.L.; Man, N.K.; Grunfeld, J.P.; Jungers, P. Factors influencing progression of renal failure in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1995, 6, 1634–1642. [Google Scholar] [PubMed]
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Kimberling, W.J.; Lezotte, D.C.; Duley, I.T.; Jones, R.H. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992, 41, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Chapman, A.B.; Torres, V.E. Volume progression in autosomal dominant polycystic kidney disease: The major factor determining clinical outcomes. Clin. J. Am. Soc. Nephrol. 2006, 1, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Shamshirsaz, A.A.; Reza Bekheirnia, M.; Kamgar, M.; Johnson, A.M.; McFann, K.; Cadnapaphornchai, M.; Nobakhthaghighi, N.; Schrier, R.W. Autosomal-dominant polycystic kidney disease in infancy and childhood: Progression and outcome. Kidney Int. 2005, 68, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Steinman, T.I. Polycystic kidney disease: A new perspective from the beginning. Kidney Int. 2005, 68, 2398–2399. [Google Scholar] [CrossRef] [PubMed]
- Reeders, S.T.; Breuning, M.H.; Davies, K.E.; Nicholls, R.D.; Jarman, A.P.; Higgs, D.R.; Pearson, P.L.; Weatherall, D.J. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 1985, 317, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.J.; Spruit, L.; Saris, J.J.; Ravine, D.; Sandkuijl, L.A.; Fossdal, R.; Boersma, J.; van Eijk, R.; Norby, S.; Constantinou-Deltas, C.D.; et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat. Genet. 1993, 5, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Igarashi, P.; Somlo, S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [PubMed]
- Tsiokas, L.; Kim, E.; Arnould, T.; Sukhatme, V.P.; Walz, G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. USA 1997, 94, 6965–6970. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Germino, F.J.; Cai, Y.; Zhang, X.; Somlo, S.; Germino, G.G. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 1997, 16, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Newby, L.J.; Streets, A.J.; Zhao, Y.; Harris, P.C.; Ward, C.J.; Ong, A.C. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 2002, 277, 20763–20773. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P.; Nomura, H.; Li, X.; Lakkis, M.; Luo, Y.; Segal, Y.; Fernandez-Fernandez, J.M.; Harris, P.; Frischauf, A.M.; Brown, D.A. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J. Biol. Chem. 2002, 277, 11276–11283. [Google Scholar] [CrossRef] [PubMed]
- Delmas, P. The gating of polycystin signaling complex. Biol. Res. 2004, 37, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A.; Spring, K.R. Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 2001, 184, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H.A.; Praetorius, J.; Nielsen, S.; Frokiaer, J.; Spring, K.R. β1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am. J. Physiol. Ren. Physiol. 2004, 287, F969–F978. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.J.; Shillingford, J.M.; Vasanth, S.; Doerr, N.; Mukherjee, S.; Kinter, M.T.; Watnick, T.; Weimbs, T. Polycystin-1 regulates STAT activity by a dual mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 7985–7990. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ding, T.; Fu, Y.; Li, C.; Cui, L.; Li, A.; Lian, P.; Liang, D.; Wang, D.W.; Guo, C. Conditional mutation of Pkd2 causes cystogenesis and upregulates β-catenin. J. Am. Soc. Nephrol. 2009, 20, 2556–2569. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; van Adelsberg, J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J. Clin. Investig. 1999, 104, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Scheffers, M.S.; van der Bent, P.; Prins, F.; Spruit, L.; Breuning, M.H.; Litvinov, S.V.; de Heer, E.; Peters, D.J. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum. Mol. Genet. 2000, 9, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.M.; Sikaneta, T.; Sullivan, B.M.; Zhang, Q.; Andreucci, M.; Stehle, T.; Drummond, I.; Arnaout, M.A. Polycystin-1 interacts with intermediate filaments. J. Biol. Chem. 2001, 276, 46544–46552. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.; De Pascalis, C.; Distefano, G.; Ducano, N.; Oldani, A.; Lanzetti, L.; Boletta, A. Regulation of the microtubular cytoskeleton by Polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration. BMC Cell Biol. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Charron, A.J.; Nakamura, S.; Bacallao, R.; Wandinger-Ness, A. Compromised cytoarchitecture and polarized trafficking in autosomal dominant polycystic kidney disease cells. J. Cell Biol. 2000, 149, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Su, X.; Nguyen, V.; Roberts, K.; Li, X.; Takakura, A.; Plomann, M.; Zhou, J. Polycystin-1 regulates actin cytoskeleton organization and directional cell migration through a novel PC1-Pacsin 2-N-Wasp complex. Hum. Mol. Genet. 2014, 23, 2769–2779. [Google Scholar] [CrossRef] [PubMed]
- Boca, M.; D’Amato, L.; Distefano, G.; Polishchuk, R.S.; Germino, G.G.; Boletta, A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3β-dependent cell cell mechanical adhesion. Mol. Biol. Cell 2007, 18, 4050–4061. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.K.; Piontek, K.; Boletta, A.; Liu, L.; Qian, F.; Xu, P.N.; Germino, F.J.; Germino, G.G. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 2002, 109, 157–168. [Google Scholar] [CrossRef]
- Lal, M.; Song, X.; Pluznick, J.L.; Di Giovanni, V.; Merrick, D.M.; Rosenblum, N.D.; Chauvet, V.; Gottardi, C.J.; Pei, Y.; Caplan, M.J. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum. Mol. Genet. 2008, 17, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.J.; Calvet, J.P. Polycystic kidney disease: In danger of being X-rated? Proc. Natl. Acad. Sci. USA 2001, 98, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Terryn, S.; Ho, A.; Beauwens, R.; Devuyst, O. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim. Biophys. Acta 2011, 1812, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Drummond, I.A. Polycystins, focal adhesions and extracellular matrix interactions. Biochim. Biophys. Acta 2011, 1812, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D.; Hreniuk, D.; Gabow, P.A. Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J. Cell. Physiol. 1992, 150, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Norman, J. Fibrosis and progression of autosomal dominant polycystic kidney disease (ADPKD). Biochim. Biophys. Acta 2011, 1812, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.P. Cyclic AMP-mediated cyst expansion. Biochim. Biophys. Acta 2011, 1812, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Somlo, G.; Chu, P.; Frankel, P.; Ye, W.; Groshen, S.; Doroshow, J.H.; Danenberg, K.; Danenberg, P. Molecular profiling including epidermal growth factor receptor and p21 expression in high-risk breast cancer patients as indicators of outcome. Ann. Oncol. 2008, 19, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Zatti, A.; Chauvet, V.; Rajendran, V.; Kimura, T.; Pagel, P.; Caplan, M.J. The C-terminal tail of the polycystin-1 protein interacts with the Na,K-ATPase α-subunit. Mol. Biol. Cell. 2005, 16, 5087–5093. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.N.; Jansson, K.; Sanchez, G.; Sharma, M.; Reif, G.A.; Wallace, D.P.; Blanco, G. Ouabain activates the Na-K-ATPase signalosome to induce autosomal dominant polycystic kidney disease cell proliferation. Am. J. Physiol. Ren. Physiol. 2011, 301, F897–F906. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.; Magenheimer, B.S.; Maser, R.L.; Calvet, J.P.; Blanco, G. Overexpression of the polycystin-1 C-tail enhances sensitivity of M-1 cells to ouabain. J. Membr. Biol. 2013, 246, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, J.; D’Agati, V.; Szabolcs, M.; Trudel, M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 1996, 13, 1153–1160. [Google Scholar] [PubMed]
- Zhou, X.J.; Kukes, G. Pathogenesis of autosomal dominant polycystic kidney disease: Role of apoptosis. Diagn. Mol. Pathol. 1998, 7, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Woo, D. Apoptosis and loss of renal tissue in polycystic kidney diseases. N. Engl. J. Med. 1995, 333, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Lager, D.J.; Qian, Q.; Bengal, R.J.; Ishibashi, M.; Torres, V.E. The pck rat: A new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001, 59, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Kim, J.; Faubel, S.; Wu, J.C.; Falk, S.A.; Schrier, R.W.; Edelstein, C.L. Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2005, 102, 6954–6959. [Google Scholar] [CrossRef] [PubMed]
- Peyronnet, R.; Sharif-Naeini, R.; Folgering, J.H.; Arhatte, M.; Jodar, M.; El Boustany, C.; Gallian, C.; Tauc, M.; Duranton, C.; Rubera, I.; et al. Mechanoprotection by polycystins against apoptosis is mediated through the opening of stretch-activated K(2P) channels. Cell Rep. 2012, 1, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Boletta, A.; Qian, F.; Onuchic, L.F.; Bhunia, A.K.; Phakdeekitcharoen, B.; Hanaoka, K.; Guggino, W.; Monaco, L.; Germino, G.G. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol. Cell 2000, 6, 1267–1273. [Google Scholar] [CrossRef]
- Wegierski, T.; Steffl, D.; Kopp, C.; Tauber, R.; Buchholz, B.; Nitschke, R.; Kuehn, E.W.; Walz, G.; Kottgen, M. TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J. 2009, 28, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Merrick, D.; Chapin, H.; Baggs, J.E.; Yu, Z.; Somlo, S.; Sun, Z.; Hogenesch, J.B.; Caplan, M.J. The γ-secretase cleavage product of polycystin-1 regulates TCF and CHOP-mediated transcriptional activation through a p300-dependent mechanism. Dev. Cell 2012, 22, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, J.; Blanco, G. Ouabain Enhances ADPKD Cell Apoptosis via the Intrinsic Pathway. Front. Physiol. 2016, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Bukanov, N. Polycystic kidney diseases: From molecular discoveries to targeted therapeutic strategies. Cell. Mol. Life Sci. 2008, 65, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, C.L. What is the role of tubular epithelial cell apoptosis in polycystic kidney disease (PKD)? Cell Cycle 2005, 4, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Mangoo-Karim, R.; Ye, M.; Wallace, D.P.; Grantham, J.J.; Sullivan, L.P. Anion secretion drives fluid secretion by monolayers of cultured human polycystic cells. Am. J. Physiol. 1995, 269, F381–F388. [Google Scholar] [PubMed]
- Albaqumi, M.; Srivastava, S.; Li, Z.; Zhdnova, O.; Wulff, H.; Itani, O.; Wallace, D.P.; Skolnik, E.Y. KCa3.1 potassium channels are critical for cAMP-dependent chloride secretion and cyst growth in autosomal-dominant polycystic kidney disease. Kidney Int. 2008, 74, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Belibi, F.A.; Wallace, D.P.; Yamaguchi, T.; Christensen, M.; Reif, G.; Grantham, J.J. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.P.; Wallace, D.P.; Grantham, J.J. Chloride and fluid secretion in polycystic kidney disease. J. Am. Soc. Nephrol. 1998, 9, 903–916. [Google Scholar] [PubMed]
- Jansson, K.; Nguyen, A.N.; Magenheimer, B.S.; Reif, G.A.; Aramadhaka, L.R.; Bello-Reuss, E.; Wallace, D.P.; Calvet, J.P.; Blanco, G. Endogenous concentrations of ouabain act as a cofactor to stimulate fluid secretion and cyst growth of in vitro ADPKD models via cAMP and EGFR-Src-MEK pathways. Am. J. Physiol. Ren. Physiol. 2012, 303, F982–F990. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.; Venugopal, J.; Sanchez, G.; Magenheimer, B.S.; Reif, G.A.; Wallace, D.P.; Calvet, J.P.; Blanco, G. Ouabain Regulates CFTR-Mediated Anion Secretion and Na,K-ATPase Transport in ADPKD Cells. J. Membr. Biol. 2015, 248, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Schaefer, C.; Krasa, H.; Oberdhan, D.; Chapman, A.; Perrone, R.D. Medical resource utilization and costs associated with autosomal dominant polycystic kidney disease in the USA: A retrospective matched cohort analysis of private insurer data. Clinicoecon. Outcomes Res. 2015, 7, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Galarreta, C.I.; Grantham, J.J.; Forbes, M.S.; Maser, R.L.; Wallace, D.P.; Chevalier, R.L. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am. J. Pathol. 2014, 184, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Brosnahan, G.; Cadnapaphornchai, M.A.; Chonchol, M.; Friend, K.; Gitomer, B.; Rossetti, S. Predictors of autosomal dominant polycystic kidney disease progression. J. Am. Soc. Nephrol. 2014, 25, 2399–2418. [Google Scholar] [CrossRef] [PubMed]
- Keenan, D.; Maxwell, A.P. Optimising the management of polycystic kidney disease. Practitioner 2016, 260, 13–16. [Google Scholar] [PubMed]
- Ferrari, P. PST 2238: A new antihypertensive compound that modulates Na,K-ATPase in genetic hypertension. J. Pharmacol. Exp. Ther. 1999, 288, 1074–1083. [Google Scholar] [PubMed]
- Ferrari, P. Rostafuroxin: An ouabain-inhibitor counteracting specific forms of hypertension. Biochim. Biophys. Acta 2010, 1802, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Ferrandi, M.; Tripodi, G.; Torielli, L.; Padoani, G.; Minotti, E.; Melloni, P.; Bianchi, G. PST 2238: A new antihypertensive compound that modulates renal Na-K pump function without diuretic activity in Milan hypertensive rats. J. Cardiovasc. Pharmacol. 2002, 40, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Bianchi, G.; Ferrari, P. Ouabain-dependent signaling in caveolae as a novel therapeutic target for hypertension. Cell. Mol. Biol. 2006, 52, 15–18. [Google Scholar] [PubMed]
- Ferrandi, M.; Manunta, P.; Ferrari, P.; Bianchi, G. The endogenous ouabain: Molecular basis of its role in hypertension and cardiovascular complications. Front. Biosci. 2005, 10, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Easterling, R.; Tietze, P.E. Digitalis toxicity. J. Am. Board Fam. Pract. 1989, 2, 49–54. [Google Scholar] [PubMed]
- Yamada, K.; Goto, A.; Hui, C.; Yagi, N.; Sugimoto, T. Effects of the Fab fragment of digoxin antibody on the natriuresis and increase in blood pressure induced by intracerebroventricular infusion of hypertonic saline solution in rats. Clin. Sci. 1992, 82, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Krep, H.; Price, D.A.; Soszynski, P.; Tao, Q.F.; Graves, S.W.; Hollenberg, N.K. Volume sensitive hypertension and the digoxin-like factor. Reversal by a Fab directed against digoxin in DOCA-salt hypertensive rats. Am. J. Hypertens. 1995, 8, 921–927. [Google Scholar] [CrossRef]
- Agunanne, E.; Horvat, D.; Uddin, M.N.; Puschett, J. The treatment of preeclampsia in a rat model employing Digibind. Am. J. Perinatol. 2010, 27, 299–305. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venugopal, J.; Blanco, G. On the Many Actions of Ouabain: Pro-Cystogenic Effects in Autosomal Dominant Polycystic Kidney Disease. Molecules 2017, 22, 729. https://doi.org/10.3390/molecules22050729
Venugopal J, Blanco G. On the Many Actions of Ouabain: Pro-Cystogenic Effects in Autosomal Dominant Polycystic Kidney Disease. Molecules. 2017; 22(5):729. https://doi.org/10.3390/molecules22050729
Chicago/Turabian StyleVenugopal, Jessica, and Gustavo Blanco. 2017. "On the Many Actions of Ouabain: Pro-Cystogenic Effects in Autosomal Dominant Polycystic Kidney Disease" Molecules 22, no. 5: 729. https://doi.org/10.3390/molecules22050729
APA StyleVenugopal, J., & Blanco, G. (2017). On the Many Actions of Ouabain: Pro-Cystogenic Effects in Autosomal Dominant Polycystic Kidney Disease. Molecules, 22(5), 729. https://doi.org/10.3390/molecules22050729