
Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
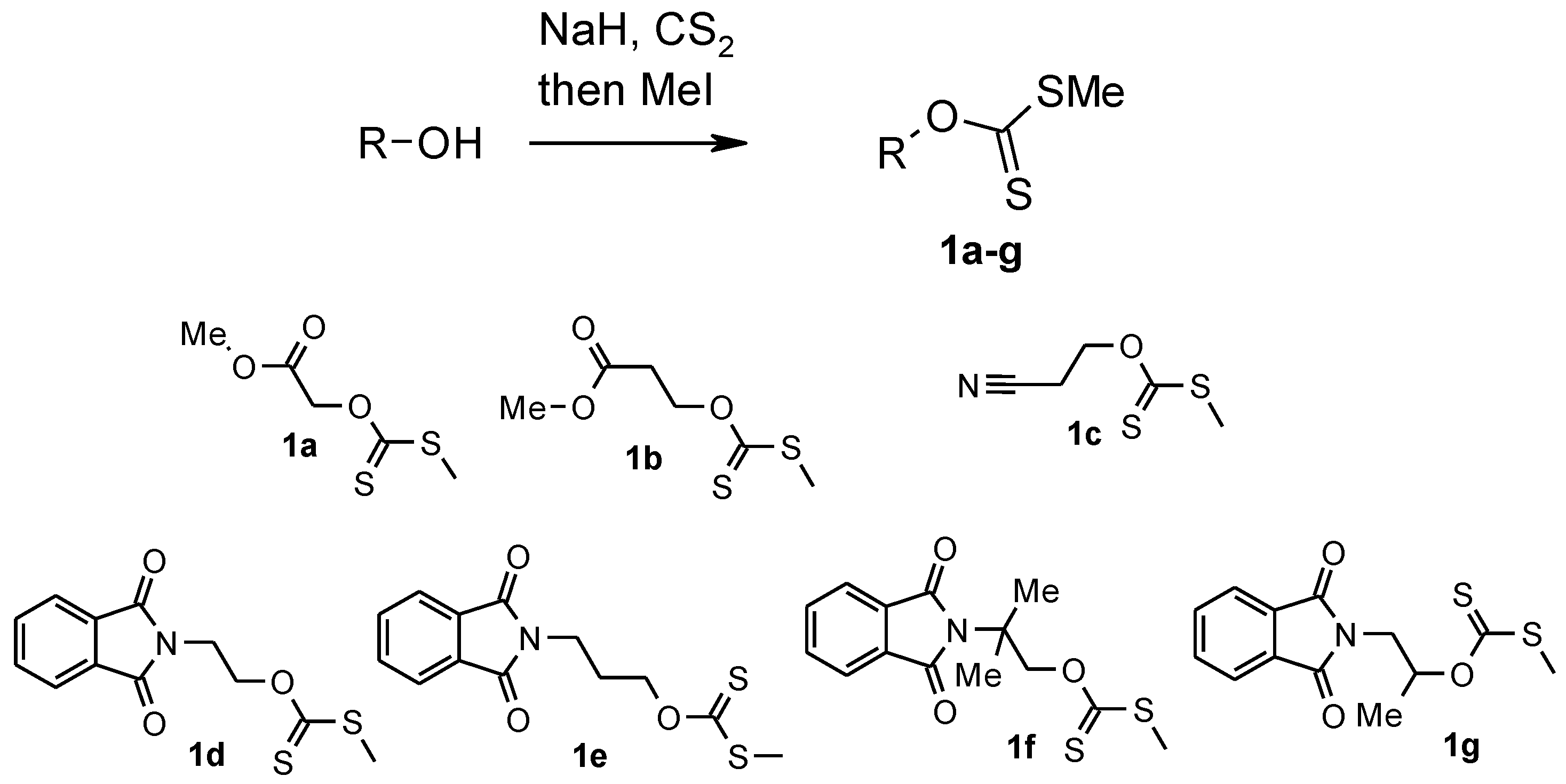
3.1. Synthesis of Xanthates 1a–c: General Procedure
3.1.1. Methyl ([(methylsulfanyl)carbonothioyl]oxy)acetate 1a
3.1.2. Methyl 3-([(methylsulfanyl)carbonothioyl]oxy)propanoate 1b
3.1.3. O-(2-Cyanoethyl)-S-methyl-dithiocarbonate 1c
3.2. Synthesis of Xanthates 1d–g: General Procedure
3.2.1. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)ethyl] S-Methyl Dithiocarbonate 1d
3.2.2. O-[3-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)propyl] S-Methyl Dithiocarbonate 1e
3.2.3. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-2-methylpropyl] S-Methyl Dithiocarbonate 1f
3.2.4. O-[2-(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)-1-methylethyl] S-Methyl Dithiocarbonate 1g
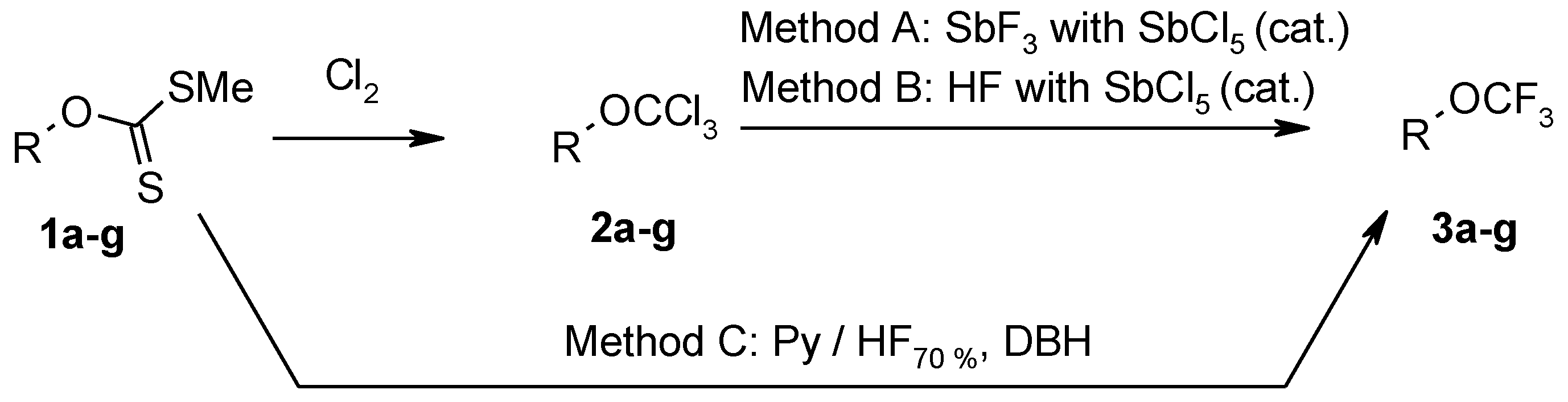
3.3. Synthesis of Trichloroderivatives 2a–g: General Procedure
3.3.1. Methyl (trichloromethoxy)acetate 2a
3.3.2. Methyl 3-(trichloromethoxy)propanoate 2b
3.3.3. 3-(Trichloromethoxy)propanenitrile 2c
3.3.4. 2-[2-(Trichloromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 2d
3.3.5. 2-[3-(Trichloromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 2e
3.3.6. 2-[1,1-Dimethyl-2-(trichloromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 2f
3.3.7. 2-[2-(Trichloromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 2g
3.4. Fluorination of Trichloromethoxy Derivatives 2a–g with Antimony Trifluoride (Method A)
3.4.1. Fluorination of Esters 2a–b and Nitrile 2c: General Procedure
3.4.2. Fluorination of Protected Amines 2d–g: General Procedure
3.5. Fluorination of Trichloromethoxy Derivatives 2a–g with Hydrogenfluoride (Method B): General Procedure
3.6. Fluorination of Xanthate 1a–g with Pyridine-Hydrogenfluoride and 1,3-Dibromo-5,5-Dimethylhydantoin (DBH) (Method C): General Procedure
3.6.1. Methyl (trifluoromethoxy)acetate 3a
3.6.2. Methyl 3-(trifluoromethoxy)propanoate 3b
3.6.3. 3-(Trifluoromethoxy)propanenitrile 3c
3.6.4. 2-[2-(Trifluoromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 3d
3.6.5. 2-[3-(Trifluoromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 3e
3.6.6. 2-[1,1-Dimethyl-2-(trifluoromethoxy)ethyl]-1H-isoindole-1,3(2H)-dione 3f
3.6.7. 2-[2-(Trifluoromethoxy)propyl]-1H-isoindole-1,3(2H)-dione 3g
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons Ltd.: Chichester, UK, 2009. [Google Scholar] [CrossRef]
- O’Hagan, D.J. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Leroux, F.; Manteau, B.; Vors, J.-P.; Pazenok, S. Trifluoromethyl ethers—Synthesis and properties of an unusual substituent. Beilstein J. Org. Chem. 2008, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased Species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef] [PubMed]
- Landelle, G.; Panossian, A.; Leroux, F.R. Trifluoromethyl Ethers and Thioethers as Tools for Medicinal Chemistry and Drug Discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Besset, T.; Jubault, P.; Pannecoucke, X.; Poisson, T. New entries toward the synthesis of OCF3-containing molecules. Org. Chem. Front. 2016, 3, 1004–1010. [Google Scholar] [CrossRef]
- Tlili, A.; Toulgoat, F.; Billard, T. Synthetic Approaches to Trifluoromethoxy-Substituted Compounds. Angew. Chem. Int. Ed. 2016, 55, 11726–11735. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Cong, F.; Guo, R.; Wang, L.; Tang, P. Asymmetric silver-catalysed intermolecular bromotrifluoromethoxylation of alkenes with a new trifluoromethoxylation reagent. Nat. Chem. 2017. [Google Scholar] [CrossRef]
- Yagupolskii, L.M. Synthesis of derivatives of phenyl trifluoromethyl ethers. Dokl. Acad. Nauk SSSR 1955, 105, 100–102, Chem. Abstr. 1956, 50, 11270b. [Google Scholar]
- Yarovenko, N.N.; Vasileva, V.S. A new method of introduction of trihalomethyl group into organic compounds. J. Gen. Chem. USSR 1958, 28, 2539. [Google Scholar]
- Yagupolskii, L.M.; Alekseenko, A.N.; Ilchenko, A.Y. Perfluoroalcoxyacetic acids and they derivatives. Sov. Prog. Chem. (Engl. Transl.), 1978, 44, 52–55. [Google Scholar]
- Farnham, W.B.; Middleton, W.J. Tris(disubstituted amino)sulfonium perfluoroalkoxides and perfluoroalkylmercaptides and process for their preparation. US Patent 4628094, 9 December 1986. [Google Scholar]
- Kanie, K.; Tanaka, Y.; Shimizu, M.; Kuroboshi, M.; Hiyama, T. Oxidative desulfurization-fluorination of alkanol xanthates. Control of the reaction pathway to fluorination or trifluoromethoxylation. Chem. Commun. 1997, 3, 309–310. [Google Scholar]
- Thibaudeau, C.; Nishizono, N.; Sumita, Y.; Matsuda, A.; Chattopadhyaya, J. Determination of the Group Electronegativity of CF3 Group in 3’-O-CF3 Thymidine by 1H-NMR. Nucleosides Nucleotides 1999, 18, 1035–1053. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Suzuki, K.; Hiyama, T. Oxidative desulfurization-fluorination of xanthates. A convenient synthesis of trifluoromethyl ethers and difluoro(methylthio)methyl ethers. Tetrahedron Lett. 1992, 33, 4173–4176. [Google Scholar] [CrossRef]
- Blazejewski, J.-C.; Anselmi, E.; Wakselman, C. 2-Trifluoromethoxyethyl Triflate: A Versatile Trifluoromethoxyethyl Carrier. J. Org. Chem. 2001, 66, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Fuse, C.; Hara, S. Synthesis of trifluoromethyl ethers and difluoro(methylthio)methyl ethers by the reaction of dithiocarbonates with IF5-pyridine-HF. J. Fluor. Chem. 2015, 179, 48–52. [Google Scholar] [CrossRef]
- Ben-David, I.; Rechavi, D.; Mishani, E.; Rozen, S. A novel synthesis of trifluoromethyl ethers via xanthates utilizing BrF3. J. Fluor. Chem. 1999, 97, 75–78. [Google Scholar] [CrossRef]
- Umemoto, T.; Singh, R.P. Arylsulfur chlorotetrafluorides as useful fluorinating agents: Deoxo- and dethioxo-fluorinations. J. Fluor. Chem. 2012, 140, 17–27. [Google Scholar] [CrossRef]
- Manteau, B.; Genix, P.; Brelot, L.; Vors, J.-P.; Pazenok, S.; Giornal, F.; Leuenberger, C.; Leroux, F.R. A General Approach to (Trifluoromethoxy)pyridines: First X-ray Structure Determinations and Quantum Chemistry Studies. Eur. J. Org. Chem. 2010, 2010, 6043–6066. [Google Scholar] [CrossRef]
- Speers, L.; Szur, A.J.; Terrell, R.C.; Treadwell, J.; Ucciardi, T.U. General anesthetics. 2. Halogenated methyl isopropyl ethers. J. Med. Chem. 1971, 14, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Nojima, M.; Kerekes, I. Synthetic Methods and Reactions II. Hydrofluorination of Alkenes, Cyclopropane and Alkynes with Poly-Hydrogen Fluoride/Pyridine (Trialkylamine)Reagents. Synthesis 1973, 1973, 779–780. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
| Entry | Chlorinated Product | Yield% | Fluorinated Product | Yield % | ||
|---|---|---|---|---|---|---|
| Method A | Method B | Method C | ||||
| 1 |  | 87 |  | traces | traces | 36 |
| 2 |  | 89 |  | 53 | 69 | 63 |
| 3 |  | 92 |  | 22 | 66 | 66 |
| 4 |  | 96 |  | 90 | 93 | 96 |
| 5 |  | 95 |  | 88 | 90 | 95 |
| 6 |  | 82 |  | 60 | 62 | 85 |
| 7 |  | 85 |  | 23 | 34 | 99 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolenko, T.M.; Dronkina, M.I.; Magnier, E.; Yagupolskii, L.M.; Yagupolskii, Y.L. Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers. Molecules 2017, 22, 804. https://doi.org/10.3390/molecules22050804
Sokolenko TM, Dronkina MI, Magnier E, Yagupolskii LM, Yagupolskii YL. Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers. Molecules. 2017; 22(5):804. https://doi.org/10.3390/molecules22050804
Chicago/Turabian StyleSokolenko, Taras M., Maya I. Dronkina, Emmanuel Magnier, Lev M. Yagupolskii, and Yurii L. Yagupolskii. 2017. "Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers" Molecules 22, no. 5: 804. https://doi.org/10.3390/molecules22050804
APA StyleSokolenko, T. M., Dronkina, M. I., Magnier, E., Yagupolskii, L. M., & Yagupolskii, Y. L. (2017). Evaluation of Efficient and Practical Methods for the Preparation of Functionalized Aliphatic Trifluoromethyl Ethers. Molecules, 22(5), 804. https://doi.org/10.3390/molecules22050804