
Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example



Abstract
:1. Introduction
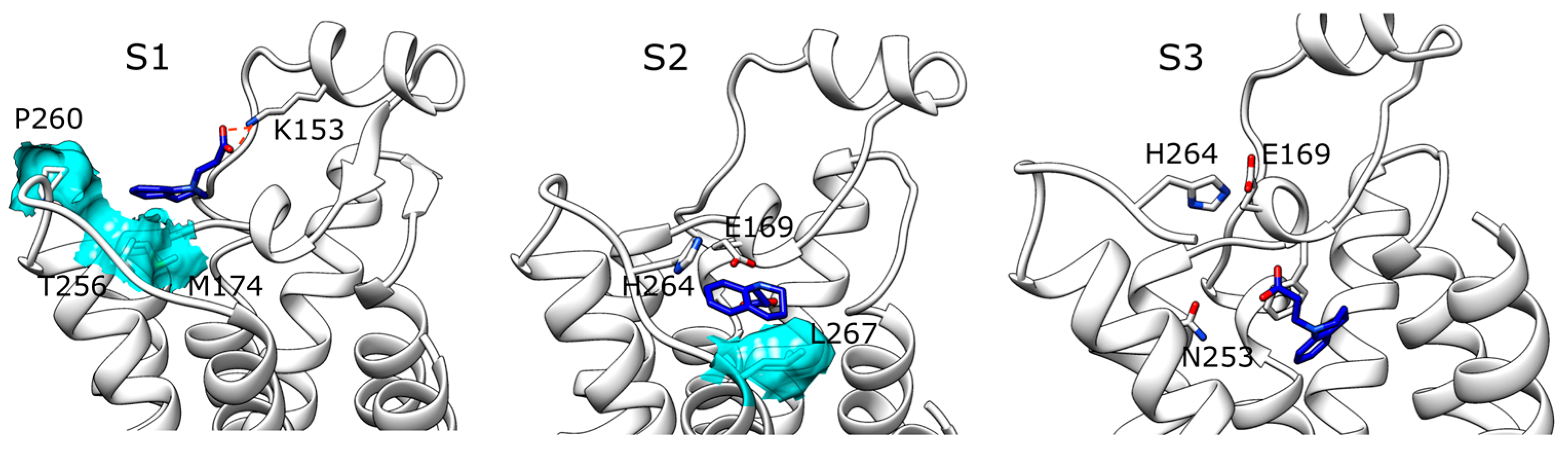
2. Results
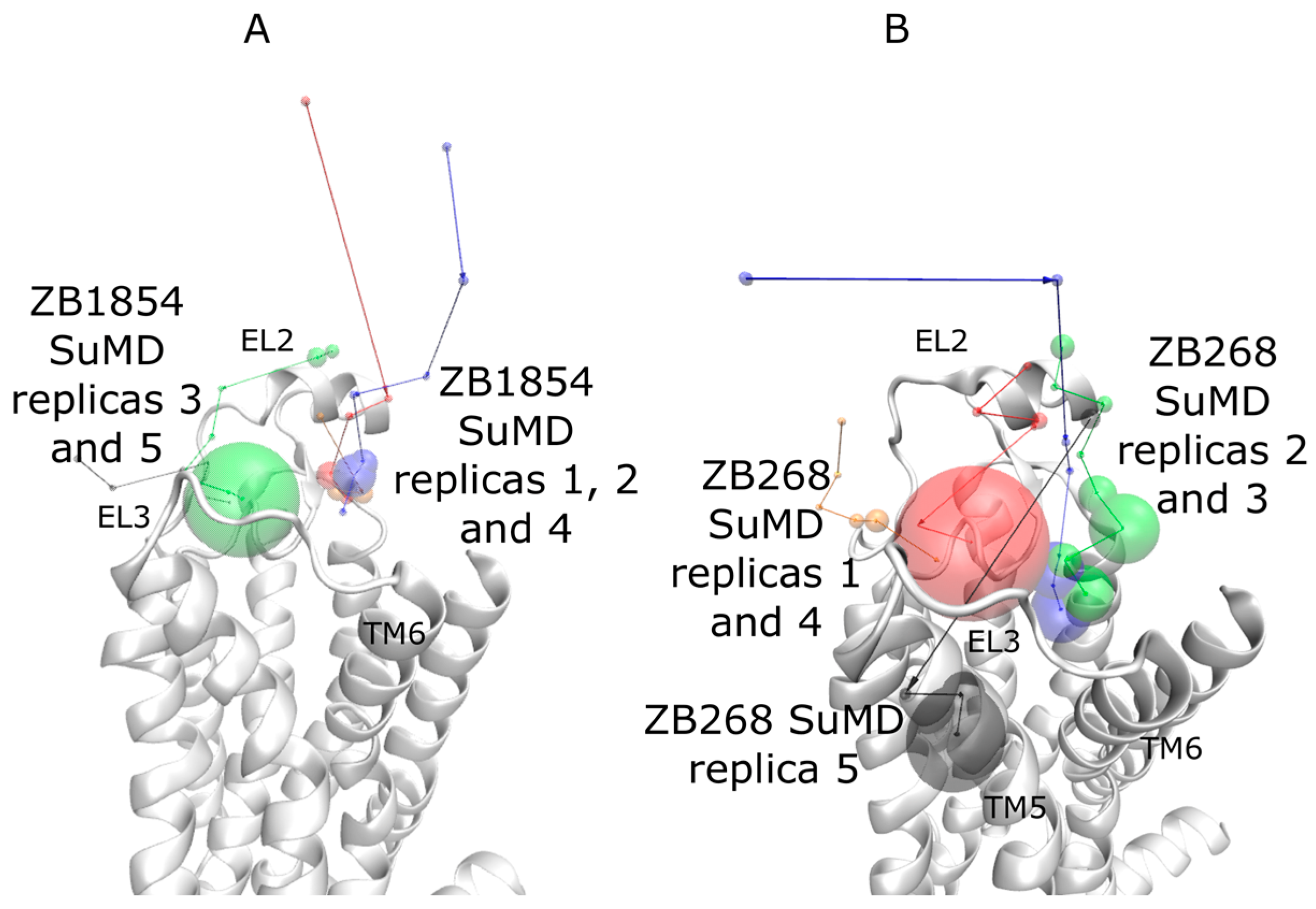
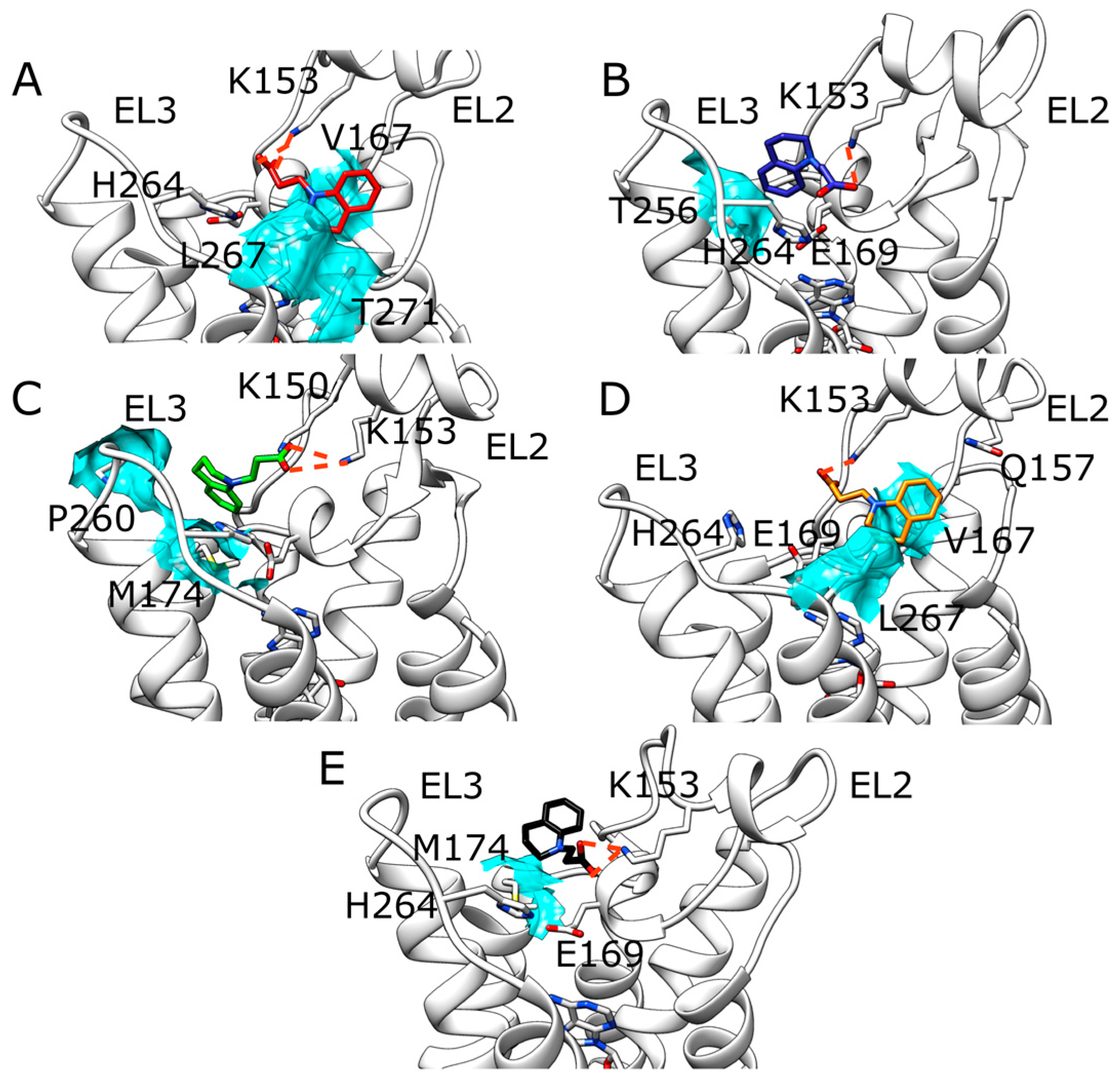
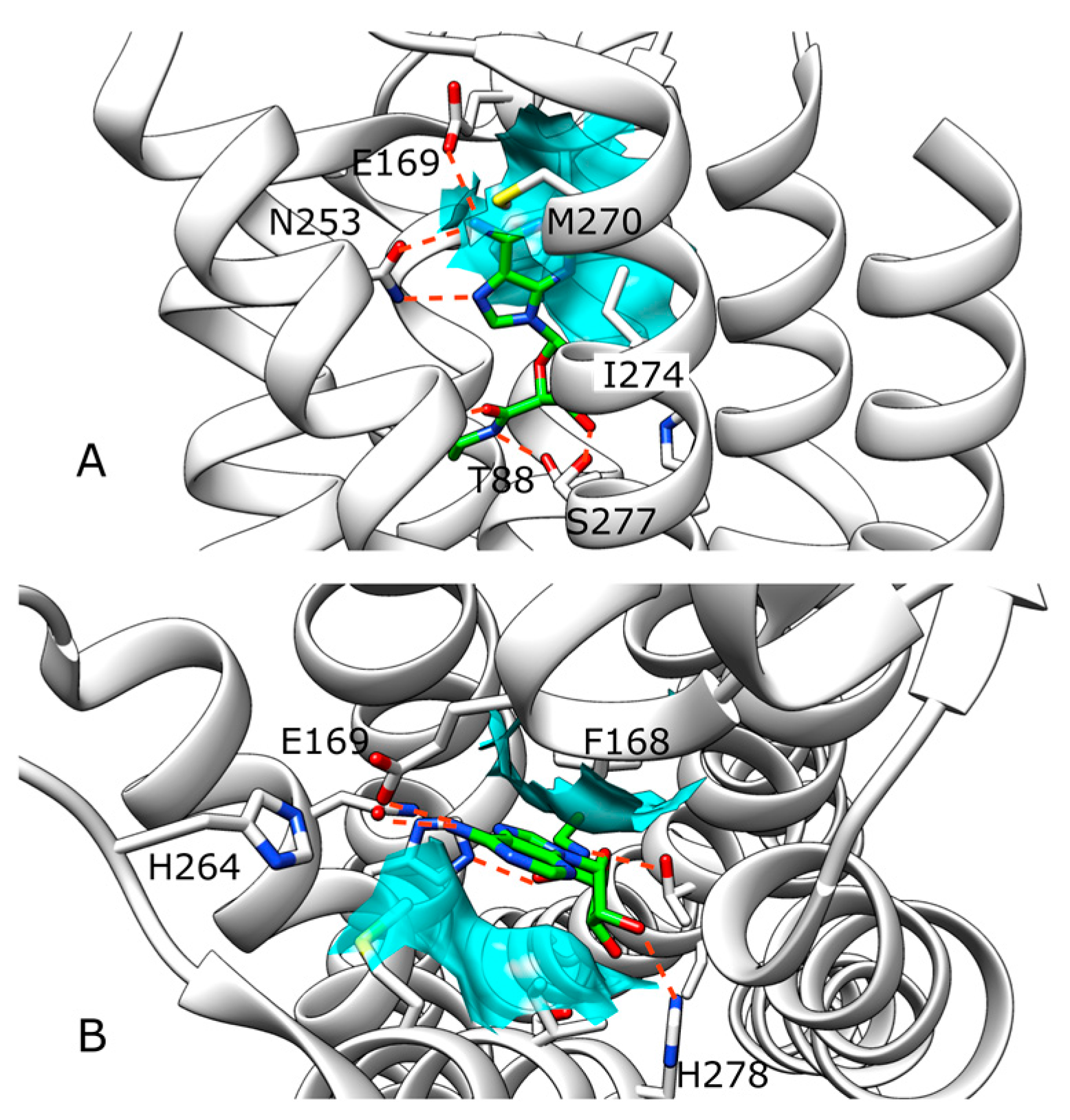
2.1. SuMD Simulations of ZB1854 on the APO Form of A2A AR
2.2. SuMD Simulations of ZB1854 on A2A AR in Orthosteric Complex with NECA
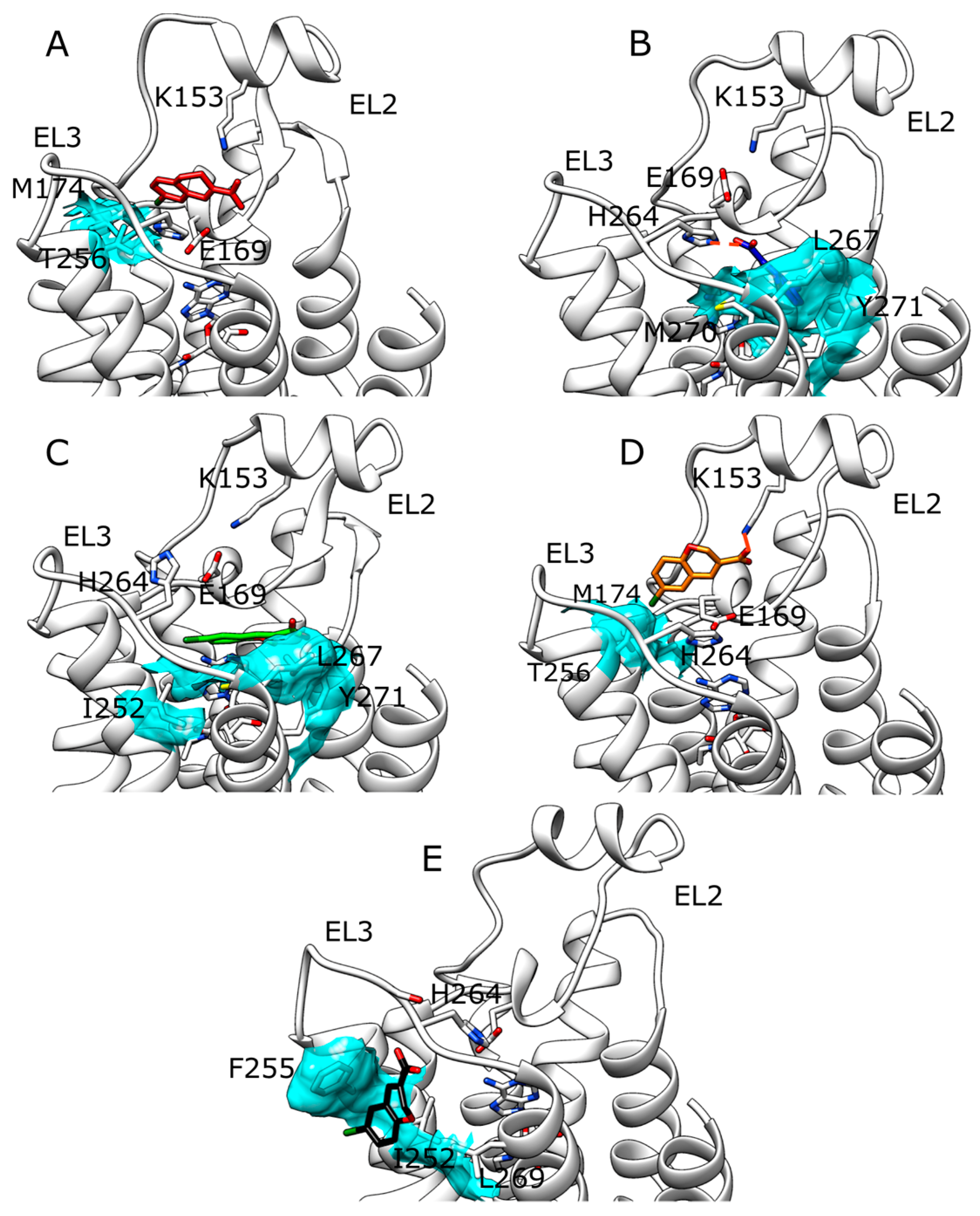
2.3. SuMD Simulations of ZB268 on A2A AR in Orthosteric Complex with NECA
3. Discussion
4. Materials and Methods
4.1. General
4.2. Systems Preparation
4.3. Ligand Parameterization
4.4. Solvated System Setup and Equilibration
4.5. Supervised Molecular Dynamics (SuMD) Simulations
4.6. Metadynamics Simulations
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

References
- Blaney, J.; Nienaber, V.; Burley, S.K. Fragment-based Lead Discovery and Optimization Using X-ray Crystallography, Computational Chemistry, and High-throughput Organic Synthesis. In Fragment-Based Approaches in Drug Discovery; Jahnke, W., Erlanson, D.A., Eds.; Methods and Principles in Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 215–248. ISBN 9783527608768. [Google Scholar] [CrossRef]
- Feyfant, E.; Cross, J.B.; Paris, K.; Tsao, D.H.H. Fragment-based drug design. Methods Mol. Biol 2011, 685, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Latham, C.F.; La, J.; Tinetti, R.N.; Chalmers, D.K.; Tachedjian, G. Fragment Based Strategies for Discovery of Novel HIV-1 Reverse Transcriptase and Integrase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 1135–1153. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, A.; Krishnamurthy, S.; Larsson, A.; Nordlund, P.; Jansson, A.; Anand, G.S. Predicting Allosteric Effects from Orthosteric Binding in Hsp90-Ligand Interactions: Implications for Fragment-Based Drug Design. PLoS Comput. Biol. 2016, 12, e1004840. [Google Scholar] [CrossRef] [PubMed]
- Lanz, J.; Riedl, R. Merging allosteric and active site binding motifs: De novo generation of target selectivity and potency via natural-product-derived fragments. ChemMedChem 2015, 10, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mou, L.; Shen, Q.; Lu, S.; Li, C.; Liu, X.; Wang, G.; Li, S.; Geng, L.; Liu, Y.; et al. ASD v2.0: Updated content and novel features focusing on allosteric regulation. Nucleic Acids Res. 2014, 42, D510–D516. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, G.; Li, S.; Liu, X.; Lu, S.; Chen, Z.; Song, K.; Yan, J.; Geng, L.; Huang, Z.; et al. ASD v3.0: Unraveling allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Res. 2016, 44, D527–D535. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Dias, J.M.; Marshall, F.H. From G Protein-coupled Receptor Structure Resolution to Rational Drug Design. J. Biol. Chem. 2015, 290, 19489–19495. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Dias, J.M.; Marshall, F.H. Structure-based drug design for G protein-coupled receptors. Prog. Med. Chem. 2014, 53, 1–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Errey, J.C.; Heitman, L.H.; Marshall, F.H.; Ijzerman, A.P.; Siegal, G. Fragment screening of GPCRs using biophysical methods: Identification of ligands of the adenosine A(2A) receptor with novel biological activity. ACS Chem. Biol. 2012, 7, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Sabbadin, D.; Federico, S.; Spalluto, G.; Moro, S. Advances in Computational Techniques to Study GPCR-Ligand Recognition. Trends Pharmacol. Sci. 2015, 36, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the strength of salt bridges: A comparison of current biomolecular force fields. J. Phys. Chem. B 2014, 118, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Gao, Z.-G.; Van Rompaey, P.; Gross, A.S.; Chen, A.; Van Calenbergh, S.; Jacobson, K.A. Modeling the adenosine receptors: Comparison of the binding domains of A2A agonists and antagonists. J. Med. Chem. 2003, 46, 4847–4859. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Gutiérrez-de-Terán, H.; Åqvist, J. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Bachhawat, P.; Chu, M.L.-H.; Wood, M.; Ceska, T.; Sands, Z.A.; Mercier, J.; Lebon, F.; Kobilka, T.S.; Kobilka, B.K. Crystal structure of the adenosine A2A receptor bound to an antagonist reveals a potential allosteric pocket. Proc. Natl. Acad. Sci. USA 2017, 114, 2066–2071. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Heitman, L.H.; IJzerman, A.P. Kinetic Aspects of the Interaction between Ligand and G Protein-Coupled Receptor: The Case of the Adenosine Receptors. Chem. Rev. 2016, 117, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Massink, A.; Gutiérrez-de-Terán, H.; Lenselink, E.B.; Ortiz Zacarías, N.V.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; IJzerman, A.P. Sodium ion binding pocket mutations and adenosine A2A receptor function. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Massink, A.; Louvel, J.; Adlere, I.; van Veen, C.; Huisman, B.J.H.; Dijksteel, G.S.; Guo, D.; Lenselink, E.B.; Buckley, B.J.; Matthews, H.; et al. 5′-Substituted Amiloride Derivatives as Allosteric Modulators Binding in the Sodium Ion Pocket of the Adenosine A2A Receptor. J. Med. Chem. 2016, 59, 4769–4777. [Google Scholar] [CrossRef] [PubMed]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G.D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group-Citing MOE. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 3 October 2016).
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Woods, R.J.; Chappelle, R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. Theochem 2000, 527, 149–156. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Grubmuller, H.; Groll, V. Solvate. Available online: http://www.mpibpc.mpg.de/grubmueller/solvate (accessed on 11 November 2015).
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ferruz, N.; De Fabritiis, G. Binding Kinetics in Drug Discovery. Mol. Inform. 2016, 35, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Moro, S. Estimation of kinetic and thermodynamic ligand-binding parameters using computational strategies. Future Med. Chem. 2017, 9, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.G.; Hoefling, M.; Aponte-Santamaría, C.; Grubmüller, H.; Groenhof, G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deganutti, G.; Moro, S. Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example. Molecules 2017, 22, 818. https://doi.org/10.3390/molecules22050818
Deganutti G, Moro S. Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example. Molecules. 2017; 22(5):818. https://doi.org/10.3390/molecules22050818
Chicago/Turabian StyleDeganutti, Giuseppe, and Stefano Moro. 2017. "Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example" Molecules 22, no. 5: 818. https://doi.org/10.3390/molecules22050818
APA StyleDeganutti, G., & Moro, S. (2017). Supporting the Identification of Novel Fragment-Based Positive Allosteric Modulators Using a Supervised Molecular Dynamics Approach: A Retrospective Analysis Considering the Human A2A Adenosine Receptor as a Key Example. Molecules, 22(5), 818. https://doi.org/10.3390/molecules22050818