
From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods


Abstract
:1. Introduction
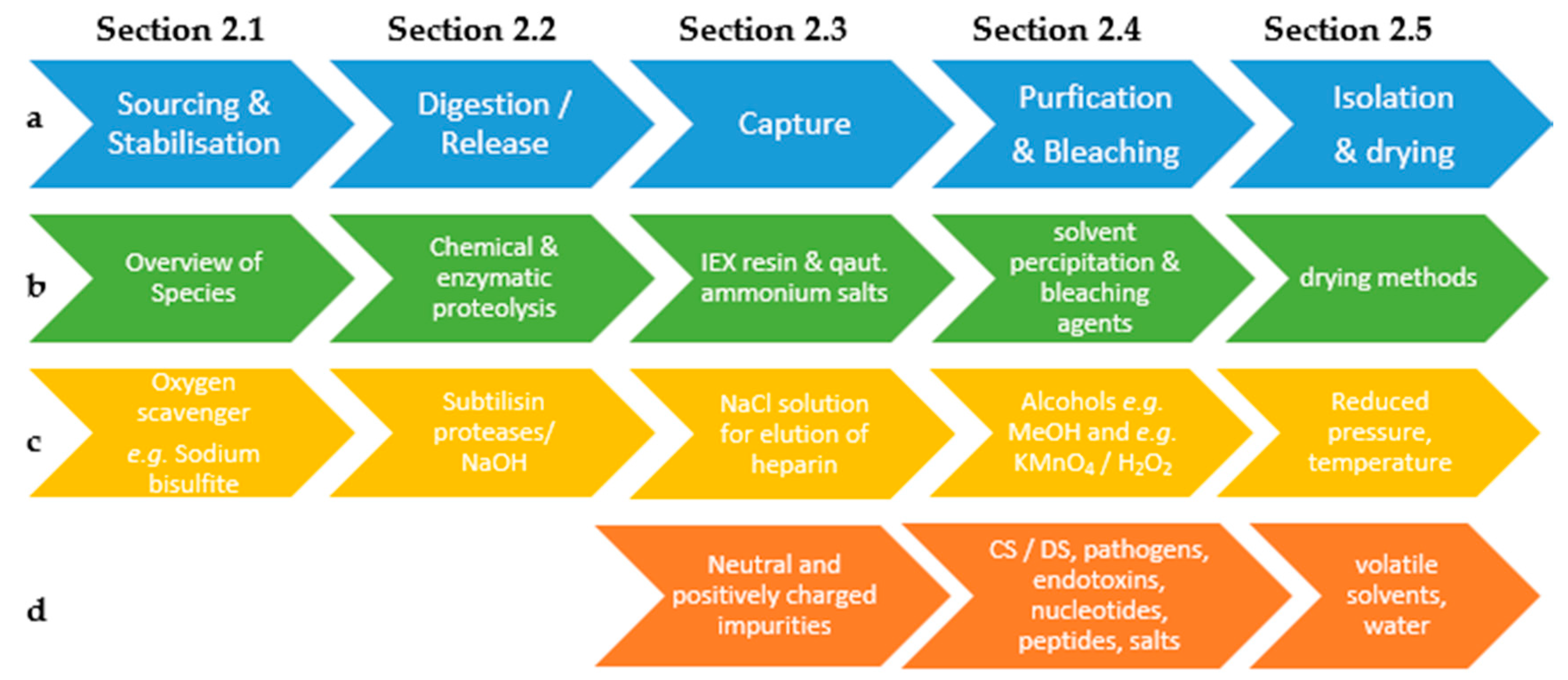
2.1. Collection and Stabilization Starting Material
2.1.1. Regulatory Aspects Related to Sourcing
2.1.2. Sources: Porcine and Bovine
2.1.3. Other Mammalian Sources
2.1.4. Non-Mammalian Sources
2.1.5. Obtaining and Stabilizing Source Material
2.1.6. Heparin on Resin
2.2. Digestion and Release of Heparin from Proteoglycans
2.3. Capture of the Heparin
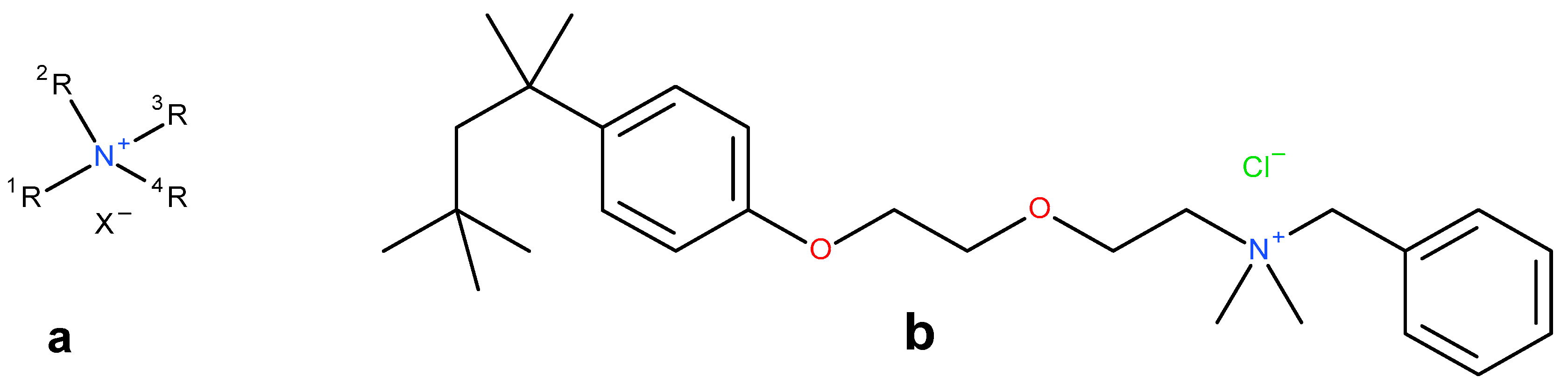
2.3.1. Precipitation with Quaternary Ammonium Salts
2.3.2. Ion Exchange Resins
2.3.3. Resins or Fractional Elution for Activity Increase
2.4. Purification and Bleaching
2.4.1. (Fractional) Precipitation
2.4.2. Bleaching
2.5. Isolation and Drying
3. Concluding Remark
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torri, G.; Naggi, A. Heparin centenary—An ever-young life-saving drug. Int. J. Cardiol. 2016, 212, S1–S4. [Google Scholar] [CrossRef]
- Yates, E.A; Rudd, T.R. Recent innovations in the structural analysis of heparin. Int. J. Cardiol. 2016, 212, S5–S9. [Google Scholar] [CrossRef]
- Barrowcliffe, T.W. History of heparin. In Heparin-A Century of Progress; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–22. [Google Scholar]
- Carlsson, P.; Kjellén, L. Heparin biosynthesis. In Heparin—A Century of Progress; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 23–41. [Google Scholar]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Jorpes, E. On the chemistry of heparin. Biochem. J. 1942, 36, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.F.; Scott, D.A. Studies on heparin: Observations on the chemistry of heparin. Biochem. J. 1936, 30, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Kruizenga, M.H.; Spaulding, L.B. The preparation of highly active barium salt of heparin and its fractionation into two chemically and biologically different constituents. J. Biol. Chem. 1943, 148, 641–647. [Google Scholar]
- Shriver, Z.; Raman, R.; Venkataraman, G.; Drummond, K.; Turnbull, J.; Toida, T.; Linhardt, R.; Biemann, K.; Sasisekharan, R. Sequencing of 3-O sulfate containing heparin decasaccharides with a partial antithrombin III binding site. Proc. Natl. Acad. Sci. USA 2000, 97, 10359–10364. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.T.; Richard, B.; Izaguirre, G.; Schedin-Weiss, S.; Gettins, P.G. Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie 2010, 92, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; Hérault, J.P.; Bernat, A.; Driguez, P.A.; Duchaussoy, P.; Lormeau, J.C.; Herbert, J.M. Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature 1999, 398, 417–422. [Google Scholar] [CrossRef] [PubMed]
- European Pharmacopeia. Monograph for Heparin Sodium Heparinum Natricum, 9th ed.; 01/2017:0333; Council of Europe: Strasbourg, France, 2017. [Google Scholar]
- Guidance for Industry-Heparin for Drug and Medical Device Use: Monitoring Crude Heparin for Quality. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291390.pdf (accessed on 19 April 2017).
- EudraLex. The Rules Governing Medicinal Products in the European Union, Volume 4: EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human Van Veterinary Use, Annex 2 Manufacture of Biological Active Substances and Medicinal Products for Human Use. Ref. Ares(2012)118531-28/06/2012. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/vol4-an2__2012-06_en.pdf (accessed on 16 June 2017).
- Keire, D.; Mulloy, B.; Chase, C.; Al-Hakim, A.; Cairatti, D.; Gray, E.; Hogwood, J.; Morris, T.; Mourão, P.A.S.; da Luz Carvalho Soares, M.; et al. Diversifying the Global Heparin Supply Chain: Reintroduction of Bovine Heparin in the United States? Pharm. Technol. 2015, 28, 36. [Google Scholar]
- Fu, L.; Li, G.; Yang, B.; Onishi, A.; Li, L.; Sun, P.; Zhang, F.; Linhardt, R.J. Structural Characterization of Pharmaceutical Heparins Prepared from Different Animal Tissues. J. Pharm. Sci. 2013, 102, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Bett, C.; Grgac, K.; Long, D.; Karfunkle, M.; Keire, D.A.; Asher, D.M.; Gregori, L. A Heparin Purification Process Removes Spiked Transmissible Spongiform Encephalopathy Agent. AAPS J. 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, P.; Liverani, L.; Mascellani, G.; Parma, B. Heterogeneity of unfractionated heparins studied in connection with species, source, and production processes. Semin. Thromb. Hemost. 1997, 23, 3–10. [Google Scholar] [CrossRef] [PubMed]
- St. Ange, K.; Onishi, A.; Fu, L.; Sun, X.; Lin, L.; Mori, D.; Zhang, F.; Dordick, J.S.; Fareed, J.; Hoppensteadt, D.; et al. Analysis of heparins derived from bovine tissues and comparison to porcine intestinal heparins. Clin. Appl. Thromb. Hemost. 2016, 22, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Hoppensteadt, D.; Maia, P.; Silva, A.; Kumar, E.; Guler, N.; Jeske, W.; Kahn, D.; Walenga, J.M.; Coyne, E.; Fareed, J. Resourcing of Heparin and Low Molecular Weight Heparins from Bovine, Ovine, and Porcine Origin. Studies to Demonstrate the Biosimilarities. Blood 2015, 126, 4733. [Google Scholar] [CrossRef]
- Warda, M.; Gouda, E.M.; Toida, T.; Chi, L.; Linhardt, R.J. Isolation and characterization of raw heparin from dromedary intestine: Evaluation of a new source of pharmaceutical heparin. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2003, 136, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Vreeburg, J.W.; Baauw, A. Method for Preparation of Heparin from Mucosa. Patent No. WO2010/110654 A1, 24 March 2009. [Google Scholar]
- Warda, M.; Mao, W.; Toida, T.; Linhardt, R.J. Turkey intestine as a commercial source of heparin? Comparative structural studies of intestinal avian and mammalian glycosaminoglycans. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2003, 134, 189–197. [Google Scholar] [CrossRef]
- Flengsrud, R.; Larsen, M.L.; Ødegaard, O.R. Purification, characterization and in vivo studies of salmon heparin. Thromb. Res. 2010, 126, e409–e417. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.P.; Paiva, J.F.; Castro, R.A.; Chavante, S.F.; Jeske, W.; Fareed, J.; Gorin, P.A.; Mendes, A.; Nader, H.B. Structural features and anticoagulant activities of a novel natural low molecular weight heparin from the shrimp Penaeus brasiliensis. Biochim. Biophys. Acta 1999, 1428, 273–283. [Google Scholar] [CrossRef]
- Cesaretti, M.; Luppi, E.; Maccari, F.; Volpi, N. Isolation and characterization of a heparin with high anticoagulant activity from the clam Tapes phylippinarum: Evidence for the presence of a high content of antithrombin III binding site. Glycobiology 2004, 14, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Z.; Linhardt, R.J. Lessons learned from the contamination of heparin. Nat. Prod. Rep. 2009, 26, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Gunay, N.S. Production and chemical processing of low molecular weight heparins. Semin. Thromb. Hemost. 1999, 25, 3–10. [Google Scholar]
- Griffin, C.C.; Linhardt, R.J.; van Gorp, C.L.; Toida, T.; Hileman, R.E.; Schubert, R.L.; Brown, S.E. Isolation and characterization of heparan sulfate from crude porcine intestinal mucosal peptidoglycan heparin. Carbohydr. Res. 1995, 276, 183–197. [Google Scholar] [CrossRef]
- Vidic, H.J. Process for Preparation of Heparin. U.S. Patent 4,283,530 A, 12 November 1976. [Google Scholar]
- Van Houdenhoven, F.A.E.; Sanders, A.L.M.; van zuthpen, P.J.J. Process for the Purification of Heparin. U.S. Patent 6,232,093, 3 January 2000. [Google Scholar]
- Homan, J.D.H.; Lens, J. A simple method for the purification of heparin. Biochim. Acta 1948, 2, 333–337. [Google Scholar] [CrossRef]
- Bush, J.A.; Freeman, S.; Hagerty, E.B. Process for Preparing Heparin. U.S. Patent 2,884,358, 22 April 1957. [Google Scholar]
- Nomine, G.; Pierre, B. Process of Purifying Heparin, and Product Produced Therefrom. U.S. Patent 2,989,438, 20 June 1961. [Google Scholar]
- Mozen, M.M.; Evans, T.D. Process for Purifying Heparin. U.S. Patent 3,058,884, 14 September 1959. [Google Scholar]
- Volpi, N. Purification of heparin, dermatan sulfate and chondroitin sulfate from mixtures by sequential precipitation with various organic solvents. J. Chromatogr. B Biomed. Sci. Appl. 1996, 685, 27–34. [Google Scholar] [CrossRef]
- Van Gorp, C.L.; Vosburgh, F.; Schubert, R.L. Protein Hydrolysate from Mucosal Tissue. U.S. Patent 5,607,840, 30 November 1992. [Google Scholar]
- Yamamoto, R.; Bellomo, E.G.; Kim, Y.; Rachana, V.Y.A.S. Method for Enhanced Heparin Quality. Patent No. WO2016/137471 A1, 26 February 2015. [Google Scholar]
- Sache, E.; Maman, M.; Bertrand, H. New Heparin fractions Having Increased Anticoagulant Activities. Patent No. GB 2,051,103 A, 10 April 1980. [Google Scholar]
- Toccaceli, N. Chromatographic Purification of Heparin. U.S. Patent 3,099,600, 30 July 1963. [Google Scholar]
- Green, J.P. Fractionation of heparin on an anion exchanger. Nature 1960, 186, 472. [Google Scholar] [CrossRef] [PubMed]
- Höök, M.; Björk, I.; Hopwood, J.; Lindahl, U. anticoagulant activity of heparin: Separation of high-activity and low-activity heparin species by affinity chromatography on immobilized antithrombin. FEBS Lett. 1976, 66, 90–93. [Google Scholar] [CrossRef]
- Volpi, N. Fractionation of heparin, dermatan sulfate, and chondroitin sulfate by sequential precipitation: A method to purify a single glycosaminoglycan species from a mixture. Anal. Biochem. 1994, 218, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Arthur, B.E.; Ernest, H.J.; William, R.L. Process of Removing Colored Impurities from Heparin. U.S. Patent 2,830,931, 28 June 1954. [Google Scholar]
- Bush, J.A.; Freeman, L.D.; Hagerty, E.B. Method for Purifying Sulfated Carbohydrates with Oxidizing Agents. U.S. Patent 31,356,660 A, 9 November 1956. [Google Scholar]
- Ewards, F.R.; Horner, A.A. Purification of Heparin. U.S. Patent 3,179,566 A, 16 May 1963. [Google Scholar]
- Celsus Website. Available online: https://www.heparin.com/manufacture_controls.php (accessed on 14 April 2017).
- Coyne, E. Heparin—Past, present and future. In Chemistry and Biology of Heparin; Lundblad, R.L., Virgil Brow, W., Mann, K.G., Roberts, H.R., Eds.; Elsevier/North-Holland: Amsterdam, The Netherlands, 1981; pp. 9–17. [Google Scholar]
- Yongjun, G. Refining Optimization Technology for Crude Heparin Sodium. Patent No. CN105,001,353 A, 17 August 2015. [Google Scholar]
- Mourier, P.; Viskov, C. Process for Oxidizing Unfractionated Heparins and Detecting Presence or Absence of Glycoserine in Heparin and Heparin Products. Patent No. WO2005090411 A1, 24 March 2004. [Google Scholar]
- Guerrini, M.; Beccati, D.; Shriver, Z.; Naggi, A.; Viswanathan, K.; Bisio, A.; Capila, I.; Lansing, J.C.; Guglieri, S.; Fraser, B.; et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol. 2008, 26, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Szajek, A.Y.; Chess, E.; Johansen, K.; Gratzl, G.; Gray, E.; Keire, D.; Linhardt, R.J.; Liu, J.; Morris, T.; Mulloy, B.; et al. The US regulatory and pharmacopeia response to the global heparin contamination crisis. Nat. Biotechnol. 2016, 34, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Szajek, A.Y.; Morris, T.S.; Koch, W.F.; Abernethy, D.R.; Williams, R.L. Inside USP: Heparin Monographs Further Revised. Pharm. Technol. 2009, 33, 136–137. [Google Scholar]
- Beccati, D.; Roy, S.; Yu, F.; Gunay, N.S.; Capila, I.; Lech, M.; Linhardt, R.J.; Venkataraman, G. Identification of a novel structure in heparin generated by potassium permanganate oxidation. Carbohydr. Polym. 2010, 82, 699–705. [Google Scholar] [CrossRef] [PubMed]
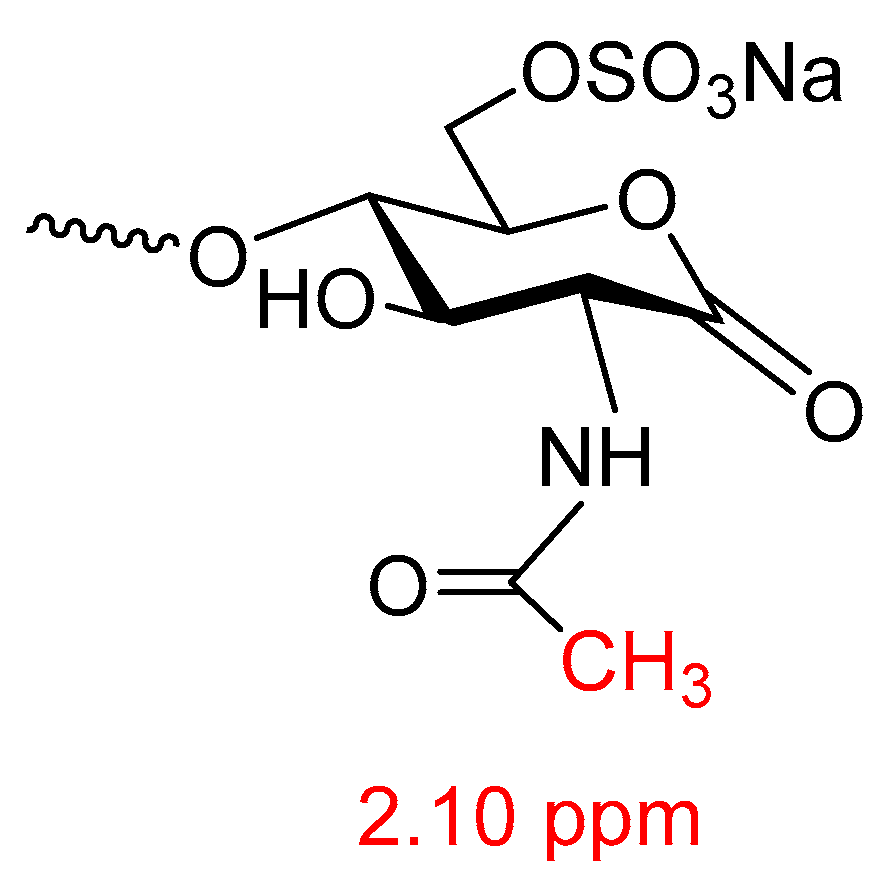
- Kellenbach, E.; Sanders, K.; Michiels, P.J.A.; Girard, F.C. 1H NMR signal at 2.10 ppm in the spectrum of KMnO4-bleached heparin sodium: Identification of the chemical origin using an NMR-only approach. Anal. Bioanal. Chem. 2011, 399, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Mourier, P.A.; Guichard, O.Y.; Herman, F.; Viskov, C. Heparin sodium compliance to the new proposed USP monograph: Elucidation of a minor structural modification responsible for a process dependent 2.10 ppm NMR signal. J. Pharm. Biomed. Anal. 2011, 54, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Gennaro, U. A conductimetric method for the determination of sulphate and carboxyl groups in heparin and other mucopolysaccharides. Carbohydr. Res. 1975, 39, 168–176. [Google Scholar] [CrossRef]
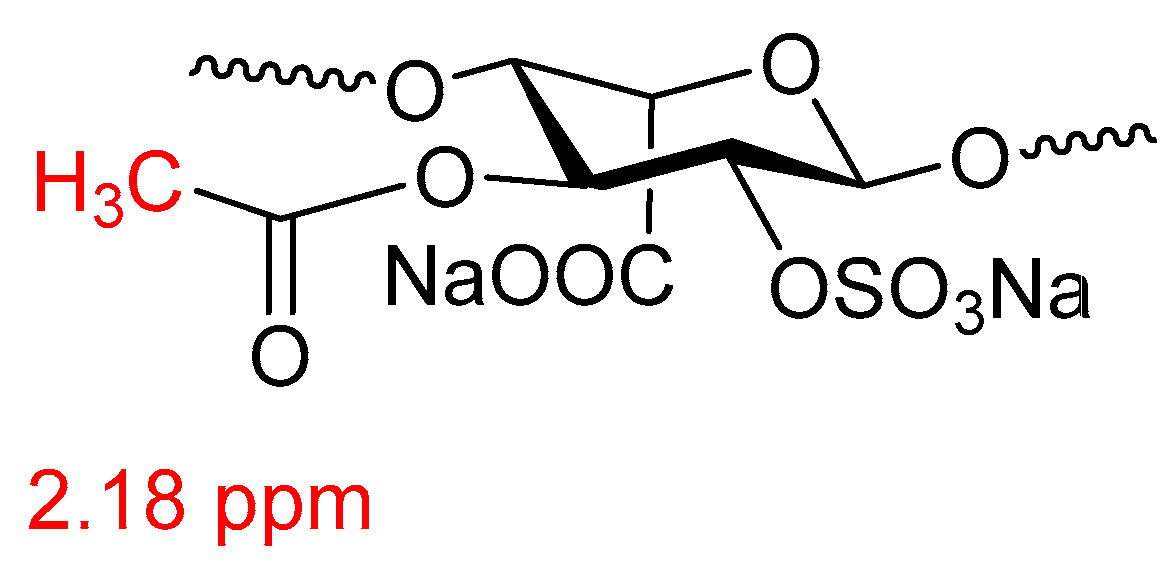
- Mourier, P.A.; Guichard, O.Y.; Herman, F.; Viskov, C. Heparin sodium compliance to USP monograph: Structural elucidation of an atypical 2.18 ppm NMR signal. J. Pharm. Biomed. Anal. 2012, 67, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Chess, E.K.; Rabinow, B.; Ray, G.J.; Szabo, C.M.; Melnick, B.; Miller, R.L.; Nair, L.M.; Moore, E.G. NMR of heparin API: Investigation of unidentified signals in the USP-specified range of 2.12–3.00 ppm. Anal. Bioanal. Chem. 2011, 399, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Jandik, K.A; Kruep, D.; Cartier, M.; Linhardt, R.J. Accelerated stability studies of heparin. J. Pharm. Sci. 1996, 85, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Linhardt, R.J.; Zhang, Z. Quantitative analysis of anions in glycosaminoglycans and application in heparin stability studies. Carbohydr. Polym. 2014, 106, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tang, J.; Wang, Z.; Zhou, J.; Yan, H. Method for Drying Heparin by Atomization. Patent No. CN1,218,058, 26 November 1997. [Google Scholar]
- Raghavan, R.; Jett, J.L. Method to Produce a Solid Form of Heparin, U.S. Patent 2004/0176581, 27 February 2004. [Google Scholar]
- WHO Model Lists of Essential Medicines. Available online: http://www.who.int/medicines/publications/essentialmedicines/EML_2015_FINAL_amended_NOV2015.pdf?ua=1 (accessed on 8 May 2017).







| Source | aXa Activity (IU/mg) | aPTT Activity | Average Mol. Weight (kDa) | S/C Ratio | Yield (mg/kg) | Refs |
|---|---|---|---|---|---|---|
| Porcine | 148–219 | 168–277 | 15.0–19.0 a | 2.31–2.57 | 160–260 | [15,16,18,22] |
| Bovine b | 123–156 | 103–181 | 16.2–16.5 | 2.29–2.40 | n.d. | [16,18,19] |
| Ovine | 142 | 150 | 22.9 | 3.66 | n.d. | [16] |
| Dromedary | 50–60 | n.d. | 24.0 | 2.0 | 400 | [21] |
| Chicken | 111 | 133 | n.d. | 2.26 | n.d. | [18] |
| Turkey | 16.6 | n.d. | n.d. | n.d. | 300 | [23] |
| Salmon | 110–137 | n.d. | <8.0 c | 2.20 | n.d. | [24] |
| Shrimp | 95–100 | n.d. | 8.5 | n.d. | 32 | [25] |
| Clam | 317 | 347 | 14.9 | n.d. | 2100 | [26] |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Meer, J.-Y.; Kellenbach, E.; Van den Bos, L.J. From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules 2017, 22, 1025. https://doi.org/10.3390/molecules22061025
Van der Meer J-Y, Kellenbach E, Van den Bos LJ. From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules. 2017; 22(6):1025. https://doi.org/10.3390/molecules22061025
Chicago/Turabian StyleVan der Meer, Jan-Ytzen, Edwin Kellenbach, and Leendert J. Van den Bos. 2017. "From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods" Molecules 22, no. 6: 1025. https://doi.org/10.3390/molecules22061025
APA StyleVan der Meer, J. -Y., Kellenbach, E., & Van den Bos, L. J. (2017). From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules, 22(6), 1025. https://doi.org/10.3390/molecules22061025