1. Introduction
Obesity is considered an important health issue since it is a risk factor for the development of a number of chronic diseases such as cardiovascular disease (primarily heart disease and stroke), diabetes, skeletal muscle disorders, and some cancers such as endometrial, breast, and colon [
1]. Human obesity is defined as a BMI (body mass index) equal to or greater than 30 kg/m
2. A person with a BMI of 25 and 30 kg/m
2 is considered overweight. Moreover, according to the World Health Organization, between 1980 and 2014, the obesity index worldwide more than doubled. Worldwide, in 2014, approximately 39% of adults (aged 18 years or older) were overweight and about 13% of the adult population was obese [
1].
The development of adipose tissue involves both hyperplasia (increased number of cells) and hypertrophy (increase in cell size). Hyperplasia is related to the proliferation and differentiation of pre-adipocytes (adipogenesis), while hypertrophy is the result of an excessive accumulation of triglycerides in existing adipocytes [
2].
Much of the knowledge about the molecular mechanisms and signals involved in adipogenesis was acquired through studies using the 3T3-L1 cell line [
3]. The differentiation of these cells comprises precisely controlled sequential stages that include cell cycle arrest and clonal expansion and differentiation (first and second stages of transcription factor activation phase) through the activation of hundreds of previously silenced genes [
4]. The exposure of confluent pre-cultured adipocytes (3T3-L1) to differentiation inducers leads to early events represented by the expression of CCAAT enhancer-binding proteins (C/EBPs), C/EBP-β, and C/EBP-δ [
4,
5]. Subsequently, the cells resume their cell cycle, undergo clonal expansion, and enter a terminal differentiation process through the activation of gamma receptors by peroxisome proliferators (PPARγ) and C/EBP-α, both key regulators in the adipogenic process [
5].
Few drugs have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of obesity, and use of these has been associated with the onset of side effects, including the development of cardiovascular diseases [
6]. Therefore, studies in various parts of the world have focused on the discovery of new sources that have anti-obesity and anti-adipogenic properties.
Several oxidants, such as hydrogen peroxide, are involved in the development of obesity. These oxidants act as signaling messengers inducing phosphorylation, oxidation, and dimerization related to the expression of C/EBP-α and PPARγ [
7]. Thus, several compounds have anti-adipogenic activity because they prevent the increase in oxidant levels [
8].
Baccharis trimera (Less.) DC is traditionally known in Brazil as “carqueja” and all parts of the plant are popularly used to produce a tonic that is consumed for body weight loss [
9].
The antimicrobial [
10], anti-inflammatory, analgesic [
11], and hypoglycemic [
12] effects of
B. trimera have been reported. In addition, high-fat diet-induced obese rats showed both decreased weight and less serum cholesterol after treatment with the methanol extract of
B. trimera than did the rats that did not receive the
B. trimera extract [
13]. Corroborating these findings, in vitro tests showed that
B. trimera methanol extract inhibits pancreatic lipase and α- and β-glucosidases [
14] whereas
B. trimera aqueous extract inhibits only glucosidases [
15].
B. trimera aqueous extract also showed antioxidant activity in the DPPH test [
16] and the total radical-trapping antioxidant parameter (TRAP) test [
17] but there are no data on
B. trimera aqueous extract evaluations using other antioxidant tests. Moreover, there are no data about the anti-adipogenic effect of
B. trimera. Thus, this study was performed to examine the antioxidant activity of
B. trimera extract, as well as the effect of
B. trimera extracts on adipogenic differentiation of 3T3-L1 cells.
3. Discussion
The presence of a large amount of reactive oxygen species (ROS) in preadipocyte cytoplasm is related to the acceleration of adipocyte differentiation through the expression of adipogenic genes in response to oxidative stress [
18]. Furthermore, antioxidant and anti-adipogenic effects of several natural compounds are known to suppress adipogenesis by preventing increased ROS levels [
8]. The decrease in excessive ROS levels and the inhibition of adipocyte differentiation are important strategies for preventing various chronic diseases related to oxidative stress and obesity. With this in mind, we first evaluated the antioxidant potential of
B. trimera extracts.
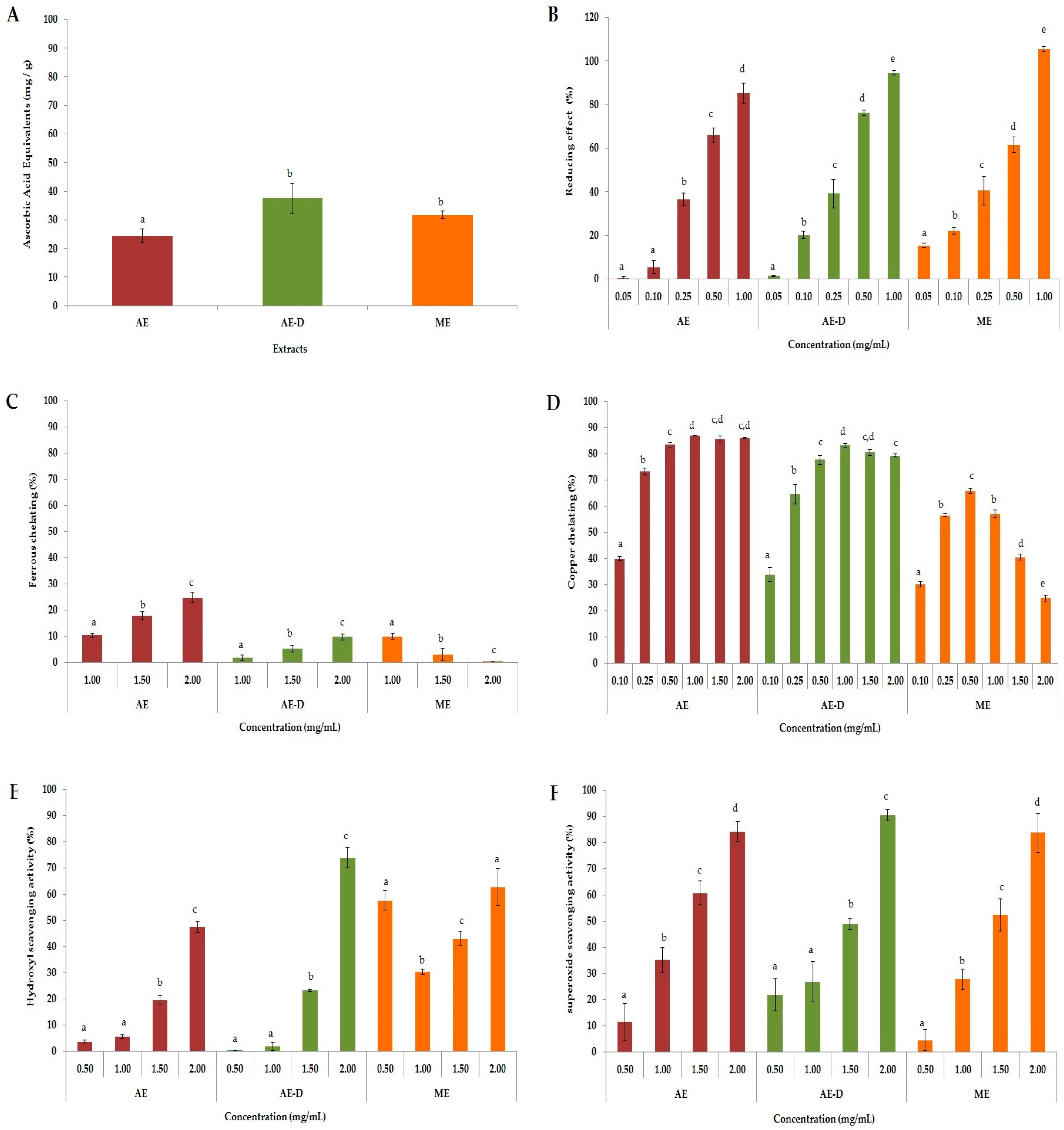
ROS are formed through a chain reaction involving three steps (initiation, propagation, and termination). Thus, different methods were used to evaluate the antioxidant properties of AE, AE-D and ME at different stages: initiation (total antioxidant capacity (TAC) and reducing power), propagation (chelation of copper and iron), and termination (scavenging of the hydroxyl superoxide radical and of hydrogen peroxide), as shown in
Figure 1.
In the TAC assay, our results were similar to those obtained by Oliveira et al. [
19]. However, our work is the first to evaluate
B. trimera extracts via a reducing power assay. Both of these tests evaluated the capacities of the samples to donate electrons and/or a hydrogen atom and break the free radical chain, however the test conditions are not similar. Therefore, when a compound acts as a donor in these two tests, it is an indication that it can act as an antioxidant in different conditions, such as in different cellular microenvironments.
All
B. trimera extracts were able to scavenge superoxide radicals in a dose-dependent manner (
Figure 1F). The superoxide radical is a highly toxic species produced by innumerable biological and photochemical reactions. This radical is a ROS that can damage various molecules, including proteins, lipids, and cellular DNA, causing cell and tissue damage as well as a number of diseases [
20]. It is produced in vivo and leads to the formation of hydrogen peroxide (H
2O
2), another oxidant agent very harmful to cells, via a dismutation reaction. Thus, by scavenging superoxide radicals, the extracts also inhibit the formation of H
2O
2.
In cells, H
2O
2 reacts with transition metal ions such as Cu
2+ and Fe
2+ to generate hydroxyl radicals via Fenton or Haber–Weiss reactions. The hydroxyl radical is the most reactive of ROS, causing serious damage to biomolecules. Its elimination is highly desirable since the elevated reactivity of this radical is associated with cellular damage, leading to several diseases [
21]. In our study, all three extracts, particularly AE-D and ME, showed hydroxyl radical scavenging activity. Vieira and Co-workers [
22] have also shown that aqueous and ethanol
B. trimera extracts were able to scavenge hydroxyl radicals. They demonstrated that the aqueous extract was a stronger scavenger than the ethanol extract, which agrees with our data. However, these authors did not address whether the plant extracts only scavenge hydroxyl radicals or if they also act as a metal chelator. Our results show that the extracts have also metal chelating activity, especially copper chelating action, which also leads to a reduction in hydroxyl radical generation.
Still with respect to the chelating activity of the extracts. We can see in
Figure 1, that all the extracts showed a much higher chelating activity of copper than the chelating activity of copper. Aqueous extracts of
B. trimera have in their composition ruthin [
11], apigenin, quercetin, and luteolin [
23]. These phenolic compounds are agents with high copper chelating capacity and low iron chelating capacity [
24,
25]. What justifies the high copper chelating activity of aqueous extracts of
B. trimera.Overall, our data showed the great potential of the
B. trimera extracts as antioxidant agents. The scavenging action of plant extracts has been correlated to the presence of phenolic compounds [
26]. Indeed, we found large amount of these compounds in the extracts (
Table 1). However, ME showed similar antioxidant activity to other extracts, although it contains fewer phenolic compounds. One possible explanation is that the different conditions of extraction allowed ME to contain large amounts of phenolic compounds that act as antioxidants. Another possibility is that the methanol extracted some compounds that were not extracted by water, and that these compounds are more powerful than those found in AE and AE-D. However, the confirmation of these hypotheses requires further investigations.
Transition metal ions including Cu
2+ and Fe
2+ can increase adipose tissue mass in rats [
27]. Furthermore, since these metals increase the expression of adipogenic transcriptional factors, they contribute to 3T3-L1 cell differentiation. Excessive ROS generation is associated with increased lipid accumulation during adipocyte differentiation [
28]. With this in mind, and after we identified the potent antioxidant activity of the extracts as both copper chelators and ROS scavengers, we decided to evaluate the anti-adipogenic activity of these extracts.
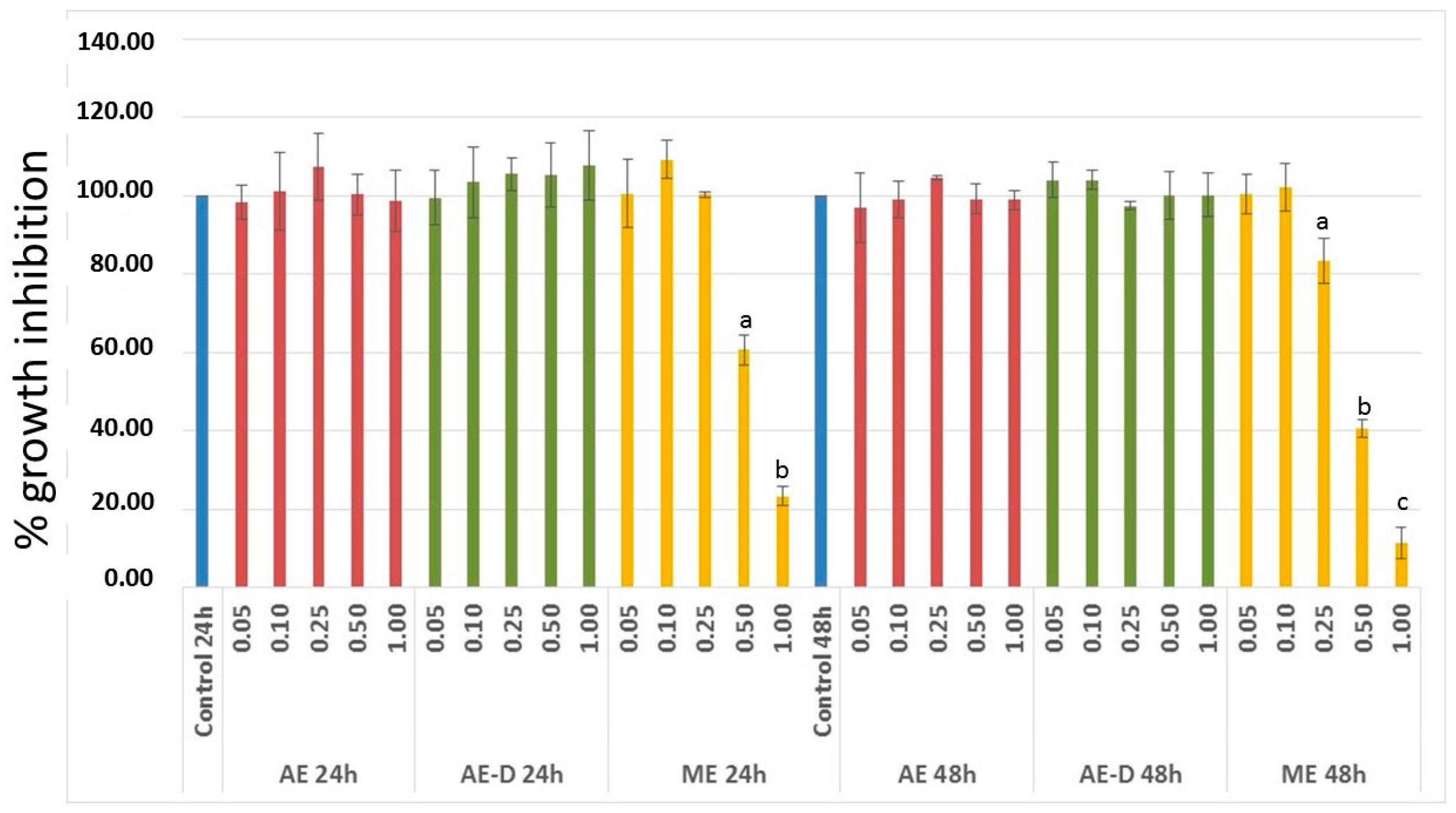
3T3-L1 cells have served as a well-documented model system used to develop anti-adipogenic agents. Therefore, it was first necessary to assess the effect of the extracts on the viability of these cells. Since ME showed anti-proliferative activity against 3T3-L1 cells (
Figure 2), it was not used in next steps of this study.
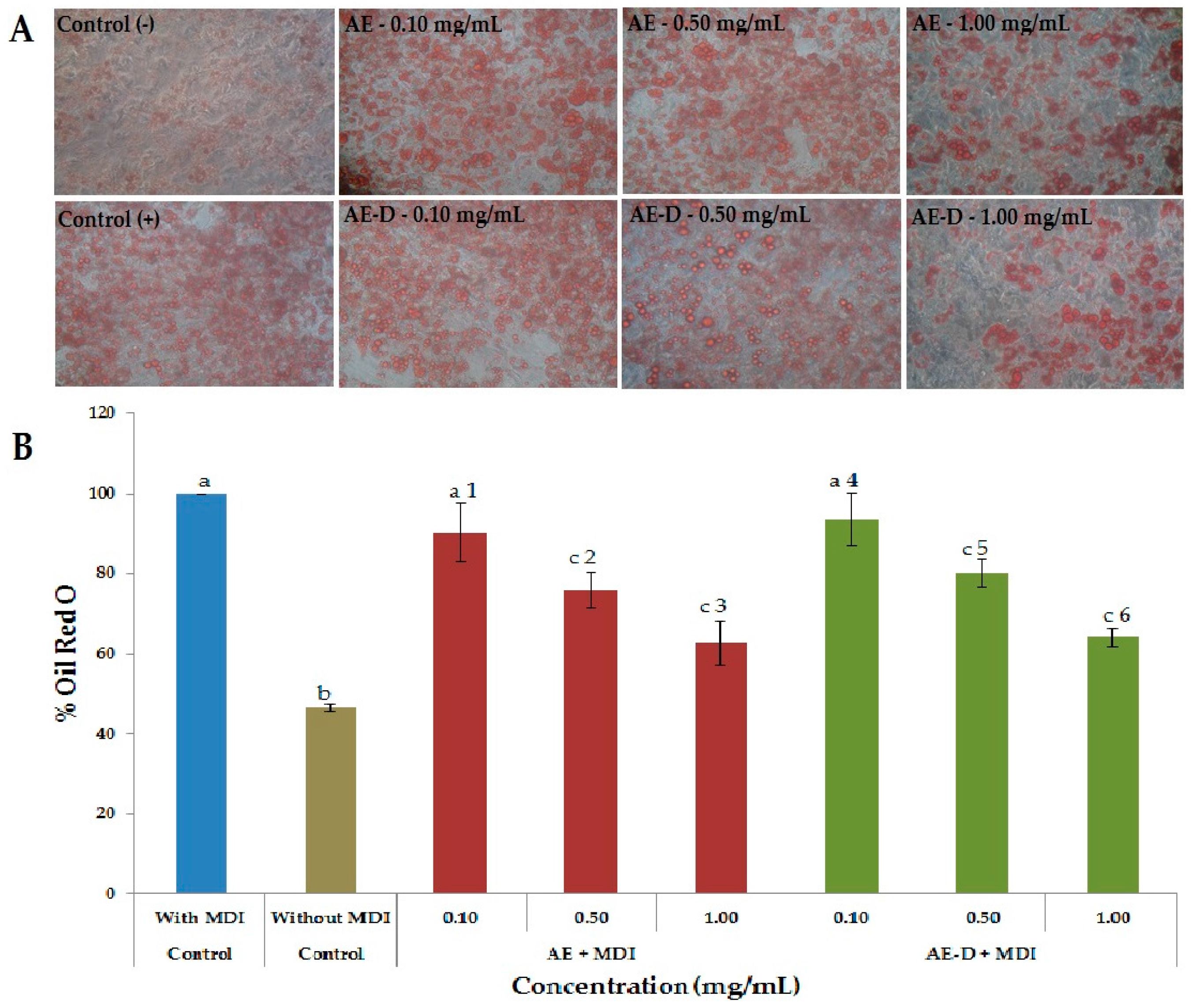
As shown in
Figure 3A,B and
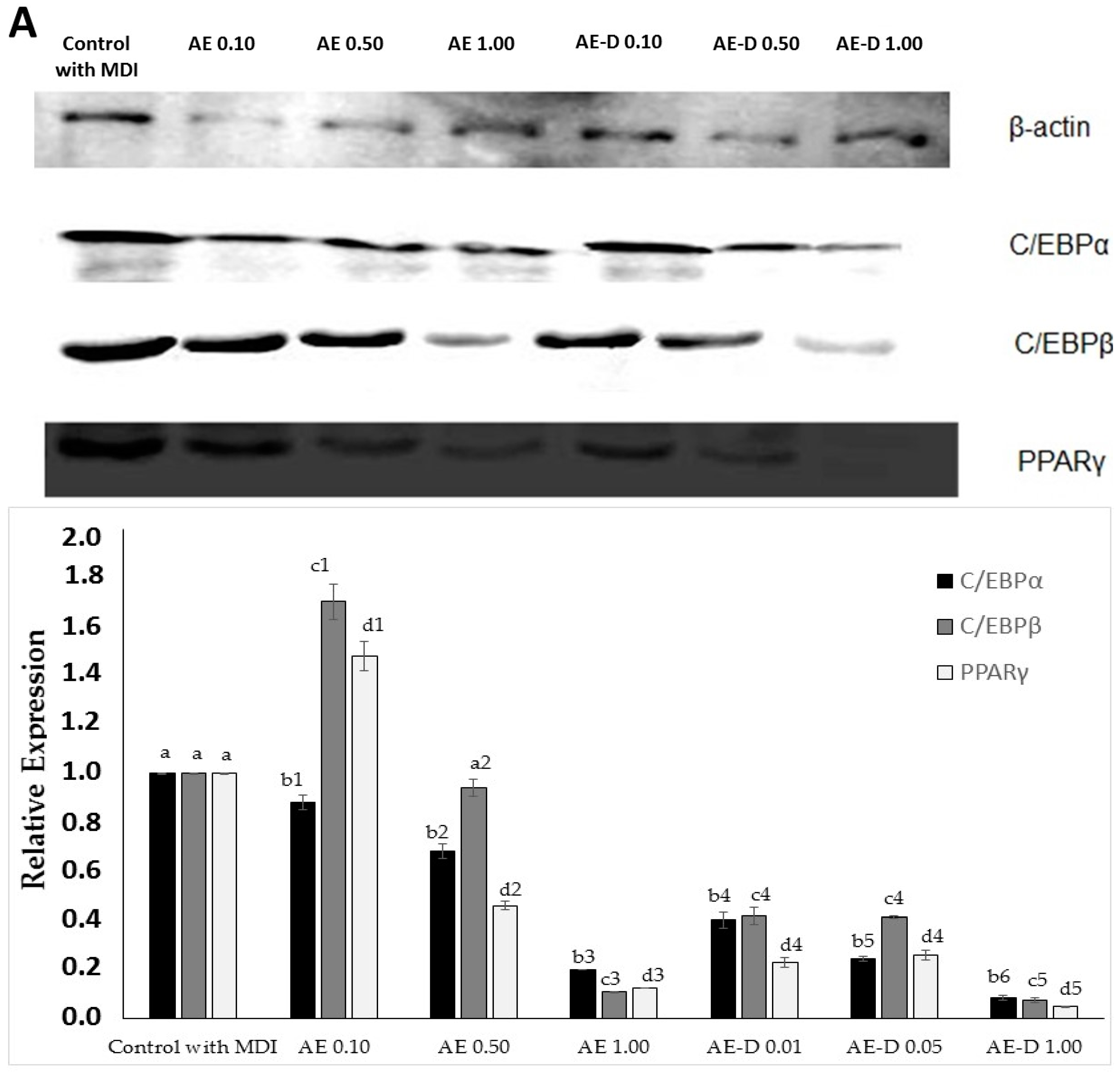
Figure 4, the AE and AE-D were able to reduce the amount of neutral lipids within the cell, a fact evidenced by the reduced staining in the cells. In order to understand their effect, we also determined the level of the transcription factors C/EBPβ, PPARγ, and C/EBPα that play key roles in the complex transcriptional cascade of adipocyte differentiation. An increase in these factors in turn leads to increased levels of molecules involved in lipid synthesis such as fatty acid synthase, fatty acid binding protein, leptin, and adiponectin [
29]. The results of the trial showed that C/EBPβ, PPARγ, and C/EBPα levels in the 3T3-L1 cells were reduced after treatment with AE and AE-D (
Figure 5).
However, these data still need to be understood. The blotting analysis showed AE and AE-D (0.10 mg/mL) at low concentration stimulates the expression of PPARγ and C/EBPβ. However, an adipogenic effect was not observed (
Figure 3 and
Figure 4), probably because the level of C/EBPα are not high. C/EBPβ is known to be a central factor in adipocyte diffusion and regulates C/EBPα levels [
4,
5]. Thus, the data obtained here lead us to propose that there are one or more compounds in the extracts that prevent the action of C/EBPβ on C/EBPα. However, the confirmation of these hypotheses requires further investigations.
B. trimera extracts have been identified as anti-obesity agents [
13,
14,
15]. However, to our knowledge, there are no other groups that have evaluated the effect of
B. trimera extracts as anti-adipogenic agents, which made a comparison of our results with those from other research groups impossible. Nevertheless, based on our data, we can say that the extracts are anti-adipogenic agents.
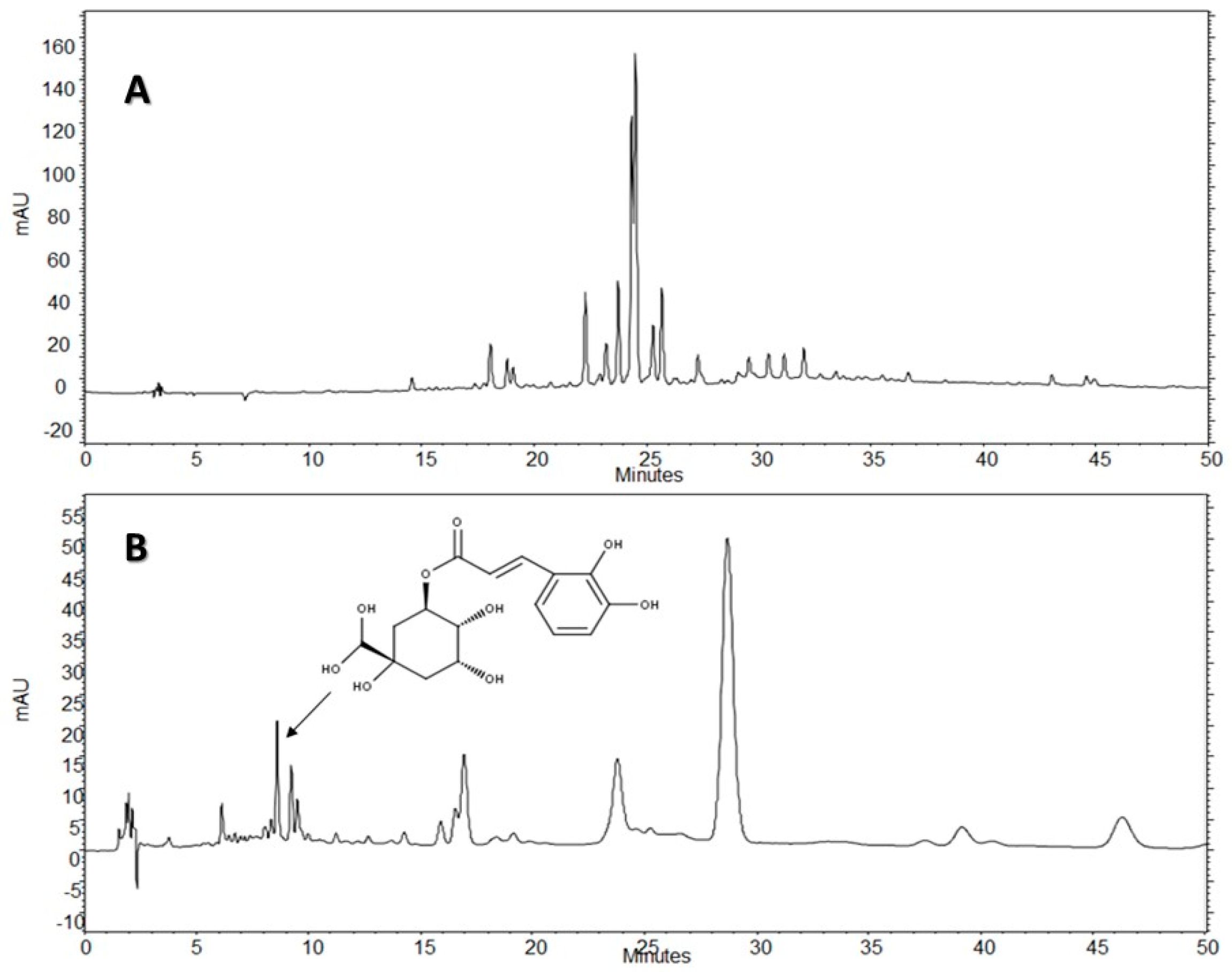
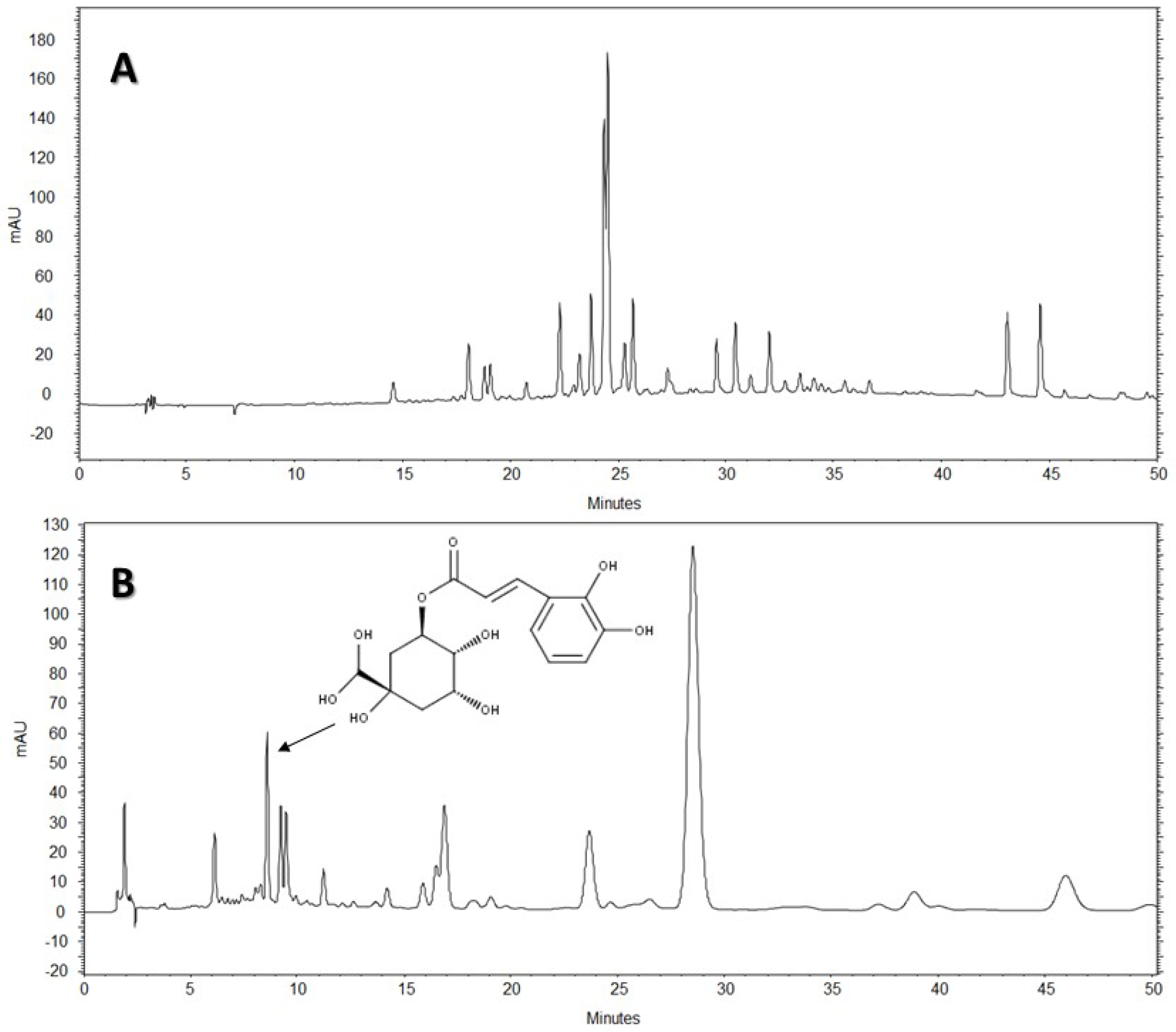
The HPLC-DAD analysis showed that AE-D and AE extracts contain several different types of phenolic compounds (
Figure 6 and
Figure 7). This result is consistent with those of previous reports. In fact, phytochemical analyses of
B. trimera extracts showed the presence of several phenolic compounds such as rutin [
11], eupafolin, hispidulin, apigenin, quercetin, and luteolin, [
23], of which these last three and rutin are possible anti-obesity agents [
9]. In our study, the data suggest the presence of CGA in
B. trimera extract composition. CGA acts as donor of hydrogen atoms to reduce free radicals and to inhibit oxidation reactions, becoming oxidized to its phenoxyl radical that is quickly stabilized by resonance stabilization as a result [
30]. Arçari and co-workers [
31] showed that CGA is a more potent anti-adipogenic compound than rutin and quercetin, since even after using a tenfold amount of these molecules, they failed to achieve the same degree of adipogenesis inhibition that they obtained using CGA. Thus, we suggested that CGA is another molecule responsible for the anti-adipogenic activity of
B. trimera extracts.
Before the all data, a question arose. Why was EA-D a more potent anti-adipogenic agent than AE? The HPLC-DAD analysis showed similar composition of these extracts, differing mainly in the concentration of the compounds. In fact, the AE extract showed the highest content of active compounds. However, AE-D was more powerful than AE in three different antioxidant tests (TAC, reducing power, and hydroxyl radical scavenging). Thus, we do not rule out the possibility that additional compound(s) present in the AE-D that are not found in the AE might also show anti-adipogenic activity. The confirmation of this hypothesis requires further investigations.
4. Materials and Methods
4.1. Materials
Potassium ferricyanide, ferrous sulfate (II), ethylenediaminetetraacetic acid (EDTA), gallic acid, ammonium molybdate, hydrogen peroxide 30%, acetic acid, Folin–Ciocalteu phenol reagent, ethanol, and sulfuric acid were obtained from Merck (Darmstadt, Germany). Nitro blue tetrazolium (NBT), monosaccharides, EDTA, ascorbic acid, methionine, bovine serum albumin (BSA), pyrocatechol violet, ascorbic acid, and ammonium molybdate were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Sodium bicarbonate, culture media components (Minimum essential Dulbecco’s modified Eagle medium (DMEM)), non-essential amino acids, fetal bovine serum, sodium pyruvate, and phosphate buffered saline (PBS) were purchased from Invitrogen Canada Inc. (Burlington, ON, Canada). Acetonitrile (HPLC grade) purchased from Panreac® (São Paulo, Brazil). Acetic acid was provided by Vetec® (São Paulo, Brazil). Water was purified with a Milli-Q system (Millipore®, Bedford, MA, USA). All other solvents and chemicals were of analytical grade.
4.2. Acquisition of B. trimera Extracts
B. trimera plants were collected in May and June 2012 in Brasilia, DF, Brazil. A specimen (No. 15694) of the plant was deposited in the Herbarium at the Department of Botany, “Universidade Federal do Rio Grande do Norte” (UFRN), Natal, RN, Brazil. The selected shoots were dehydrated in an aerated oven at 45 °C for three days and then crushed. From the ground material three different extracts were prepared according as described above.
Approximately 100 g of crushed material was boiled in 250 mL of water for 10 min and filtered, concentrated under reduced pressure at −80 °C, and lyophilized. The sample obtained was named AE-D.
ME was obtained after ground material (~100 g) was macerated into methanol (250 mL) for 24 h, at room temperature (~22 °C), under stirring for 24 h (three replicates). The mixture was then filtered, concentrated under reduced pressure at −80 °C, and lyophilized.
In order to obtain the aqueous extract (AE), the ground material (~100 g) was macerated into 250 mL of distillated water at room temperature under stirring for 24 h. The mixture was then filtered, concentrated under reduced pressure at −80 °C, and lyophilized.
4.3. Chemical Analysis
Total sugar, protein, and phenolic compound contents were determined as described previously [
32]. Phenolic compounds were determinate via the Folin-Ciocalteu phenol reagent method using gallic acid as a standard. The amounts of protein were measured using the Coomassie Brilliant Blue reagent, with BSA as a standard. Total amounts of carbohydrates were determined using the phenol-H
2SO
4 method with
d-glucose as a standard.
4.4. HPLC-DAD Profile of AE and AE-D Extracts from the Leaves of B. trimera
HPLC-DAD analyses were performed using an HPLC Merck-Hitach® (Hichrom model) instrument equipped with a diode array detector, quaternary pump, oven column, and autoinjector. For the analysis of the compounds, two methods were performed using a Phenomenex® Luna RP-18 column (250 × 4.6 mm, 5 µm particle size) and the eluents were: (A) acetic acid 0.3% and (B) acetonitrile. The following gradient (v/v) was applied: 0–50% B, 0–50 min for Method 1, and 5–10% B, 0–5 min; 10% B, 5–50 min for Method 2. Both methods had a total analysis time of 50 min. Flow elution was 1.0 mL/min for Method 1 and 1.5 mL/min for Method 2, and the injection volume was 20 µL for both methods. Detection was performed at 254, 280, and 340 nm, with the acquisition of UV spectra in the range of 200 to 400 nm.
The AE of B. trimera (heated and unheated) was compared against standards of CGA (≥95%), ellagic acid (≥95%), gallic acid (≥95%), vitexin (≥95%), all isovitexin (≥98%), all acquired from Sigma-Aldrich®. The AE of B. trimera and standards were mixed in methanol:water, 1:1 (v/v). The final concentration of the extract was 3.0 mg/mL. For standards, the final concentration was 100 μg/mL. In this technique, the identification of compounds was based on a comparison of retention times, UV spectra of the major peaks, and the increase in peak area after co-injection of standards and extracts. Each trial was performed in triplicate. After total dissolution and prior to analysis, the samples and solvents were filtered through a 0.45 μm membrane (MillexTM, Merck®).
4.5. Antioxidant Activity
The antioxidant activities of
B. trimera extracts were measured using the methods described by Melo et al. (2014) [
33], and/or Melo-Silveira et al. (2014) [
34].
4.5.1. Determination of Total Antioxidant Capacity
The assay used was based on the reduction of Mo6+ to Mo5+ by the samples and the subsequent formation of a green phosphate-molybdate complex at an acidic pH. Tubes containing extracts and reagent solution (0.6 M sulfuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate) were incubated at 95 °C for 90 min. After the mixture had cooled to room temperature, the absorbance of each solution was measured at 695 nm against a blank. The TAC was measured in ascorbic acid mg/extract g, termed equivalent of ascorbic acid.
4.5.2. Reducing Power
The reducing power assay depends on the reduction of the potassium ferricyanide by the samples. Briefly, the B. trimera extracts were mixed with a phosphate buffer 0.2 M (pH 6.6), potassium ferricyanide (1% m/v) and incubated at 50 °C for 20 min. One addition of trichloroacetic acid (10% m/v) was used to in order to stop the reaction. Distilled water and ferrous chloride (0.1% m/v) were added to the solution and the absorbing capacities were measured at 700 nm. Results were calculated as an activity percentage, considering the largest concentration of ascorbic acid (the standard) as 100% activity.
4.5.3. Ferric Chelating
The chelating ability of samples was evaluated as described below: Each extract at different concentrations was added to the reaction mixture containing FeCl2 (0.05 mL, 2 mM) and ferrozine (0.2 mL, 5 mM). The mixture was shaken and incubated for 10 min at room temperature and absorbance of the mixture was measured (562 nm) against a blank. EDTA was used as positive control.
The chelating effect was calculated using the corresponding absorbance (A) in the formula given below, where control is the absorbance in the absence of chelating agents:
4.5.4. Copper Chelation
The test was carried out in 96-well microplates with 0.2 mL of reaction mixture containing varying amount of
B. trimera extracts, pyrocatechol violet (4 mM), and copper II sulfate pentahydrate (50 mg/mL). All wells were stirred and the solution absorbance was measured at 632 nm. The control group was obtained using the same reagents in the absence of chelating agents. The ability of the
B. trimera extracts to chelate the copper ion was determinate using the following formula:
4.5.5. Hydroxyl Radical Scavenging Activity Assay
The Fenton’s reaction (Fe2+ + H2O2→Fe3+ + OH− + OH˙) was used in order to determinate the scavenging activity of B. trimera extracts against the hydroxyl radical. These data were expressed as inhibition rates. Hydroxyl radicals were generated mixing 3 mL sodium phosphate buffer (150 mM, pH 7.4), 10 mM FeSO4·7H2O, 10 mM EDTA, 2 mM sodium salicylate, and 30% H2O2 (200 mL) and varying extract concentrations. In the control, sodium phosphate buffer was replaced by H2O2. The solutions were incubated at 37 °C for 1 h, and the presence of OH− was detected by monitoring absorbance at 510 nm. Gallic acid was used as the positive control.
4.5.6. Superoxide Radical Scavenging Activity Assay
This test depends on the ability of samples to inhibit the photochemical reduction of nitroblue tetrazolium (NBT) in the riboflavin–light–NBT system. Each 3 mL of reaction mixture contained 75 mM NBT, 50 mM phosphate buffer (pH 7.8), 2 mM riboflavin, 13 mM methionine, 100 mM EDTA, and 1 mL sample solution. After the production of blue formazan the increase in absorbance at 560 nm after 10-min illumination from a fluorescent lamp was determined. The entire reaction assembly was enclosed in a box lined with aluminum foil. An identical reaction was maintained in the dark and used as a blank. The positive control was the Gallic acid. Results were accounted in scavenging percentage.
4.6. Cell Culture, Differentiation and BrdU Assay
3T3-L1 preadipocyte cells were purchased from a cell bank in Rio de Janeiro, RJ, Brazil (CR089-BCRJ/UFRJ) and maintained with 10% FBS/DMEM containing 4.5 g/L glucose, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 0.25 mg/mL amphotericin B at 37 °C in 5% CO2 incubator. Confluent cells were differentiated by incubation with the hormone mixture MDI containing 10 µg/mL insulin, 1 μM dexamethasone, and 0.5 mM IBMX, in 10% FBS/DMEM for 72 h. Thereafter, the cells were maintained in post-differentiation medium containing 10 g/mL insulin in 10% FBS/DMEM in the presence of various concentrations (0.05; 0.10; 0.25; 0.50 and 1.00 mg/mL) of B. trimera extracts and the medium was replaced every 3 days. The same concentration of B. trimera extract was supplemented at 3-day intervals when culture medium was replaced. Differentiation, as measured by the expression of adipogenic markers and appearance of lipid droplets, was completed at Day 15 as measured with the use of the dye Oil Red O.
Anti-proliferative testing was conducted using 5-Bromo-2-Deoxyuridine (BrdU) assay, as described by Nobre et al. 2014 [
35]. 3T3-L1 cells were seeded into 96-well plates at a density of 5 × 10
3 cells/well and allowed to attach overnight in 250 μL of fresh medium at culture conditions (24 h, at 37 °C and 2.5% CO
2). After 12 h, the medium was removed and the extracts B in medium was added to a final concentration of 0.05–1.0 mg/mL and incubated for 24 or 48 h. After incubation, traces of extracts were removed by washing the cells twice with 200 μL PBS and the BrdU incorporation was determined according to the manufacturer’s instruction (BrdU cell proliferation assay kit-Cell Signaling, Danvers, MA, USA).
4.7. Oil Red O Staining and Free Glycerol
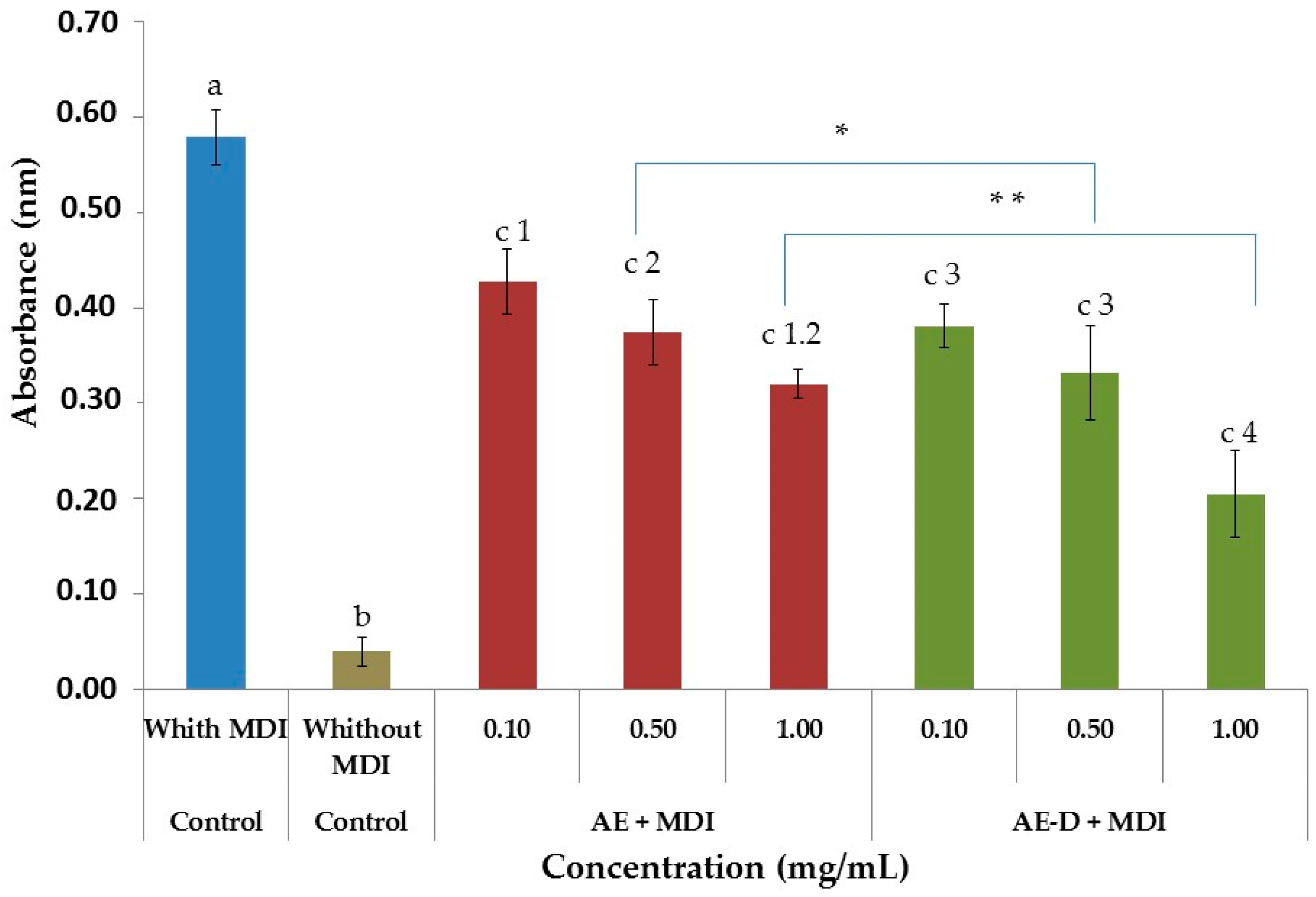
At day 15 after the induction of differentiation, cells were stained with Oil Red O. Cells were washed twice with PBS and fixed with 3.7% formaldehyde for 10 min. Fixed cells were stained with 0.2% Oil Red O-isopropanol for 1 h and the excess of stain was washed by 70% ethanol and water. Cells were then photographed using microscope (Nikon EclypseTi-E). Stained oil droplets was dissolved with isopropanol and quantified by spectrophotometric analysis at 510 nm. The optical density in only-MDI-treated cells was taken as 100% of relative lipid content. Results were represented as relative lipid contents of each experimental group.
To determine free glycerol, we used a Sigma kit (Sigma-Aldrich Co., St. Louis, MO, USA). The free glycerol reagent was made up according to the manufacturer’s instructions at room temperature. For the test were prepared solution without samples (10 µL water + 400 µL reagent), the standard (5 µL glycerol + 400 µL reagent) and samples (10 µL sample + 400 µL reagent). To complete mixing of the components, shaken by inversion and led to heating in a water bath at 37 °C for 5 min. The absorbance of the mixture was measured at 540 nm for the quantitative enzymatic determination of glycerol. The blank was used as negative control and the standard as positive. The procedure involves enzymatic hydrolysis by lipase of the triglycerides to glycerol and free fatty acids. The increase in absorbance at 540 nm is directly proportional to the free glycerol concentration of the sample.
4.8. Western Blotting Analysis
3T3-L1 cells were plated in 100 mm plates and stimulated to differentiate into adipocytes (as described above) in the presence or absence of the sample. After 15 days of differentiation, cells were removed from the plates (two plates per treatment) and subjected to washing in ice cold PBS and centrifugation. To obtain the total cellular protein extract, the cells were re-suspended in lysis buffer (50 mM Tris-HCl (pH 7.4); 1% Tween 20; 0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EGTA; 1 mM Na3VO4; 1 mM NaF; and protease inhibitors) for 2 h on ice.
After removal of cellular debris by centrifugation, the protein concentration of the extracts was determined using Bradford reagent (Bio-Rad, Hercules, CA, USA). The extracts were re-suspended in sample buffer (100 mM Tris-HCl (pH 6.8), 200 mM dithiothreitol, 4% sodium dodecyl sulfate (SDS), 0.1% bromophenol blue and 20% glycerol), and these solutions were boiled for 5 min. For each sample, approximately 30 μg of protein was subjected to SDS-polyacrylamide gel electrophoresis (PAGE) with subsequent transfer to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked by a solution containing nonfat milk (1%) or albumin (1%) in tris-buffered saline (0.05%) and Tween 20 (TBST buffer) and incubated 12 h at 4 °C with appropriate primary antibody at a dilution of 1:1000. After further washing in TBST buffer, the membranes were incubated with appropriate secondary antibody (anti-mouse or anti-rabbit) conjugate with peroxidase (dilution 1: 1000) in blocking buffer for 1 h. Detection was performed using chemiluminescence.
The bands in the gel were quantified by densitometry and a ratio between the values obtained for the C/EBPα, C/EBPβ, and PPARγ proteins and the value obtained for actin was used to construct
Figure 5.
The primary antibodies used specific for β-actin (sc-130656) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); C/EBPα (8178), C/EBPβ (3087), and PPARγ (2443) were obtained from Cell Signaling Technology (Beverly, MA, USA). The horseradish peroxidase (HRP) Secondary antibodies were also obtained from Cell Signaling Technology (Beverly, MA, USA).
4.9. Statistical Analysis
All experiments were repeated at least three times, and all values are given as mean ± standard deviation (S.D.) of triplicates. One-way analysis of variance (ANOVA) followed by a Student’s t-test was used to analyze statistical significance. Results with p < 0.05 were considered as statistically significant.



 and
and 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}