Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies


Abstract
:1. Introduction
2. Results and Discussion
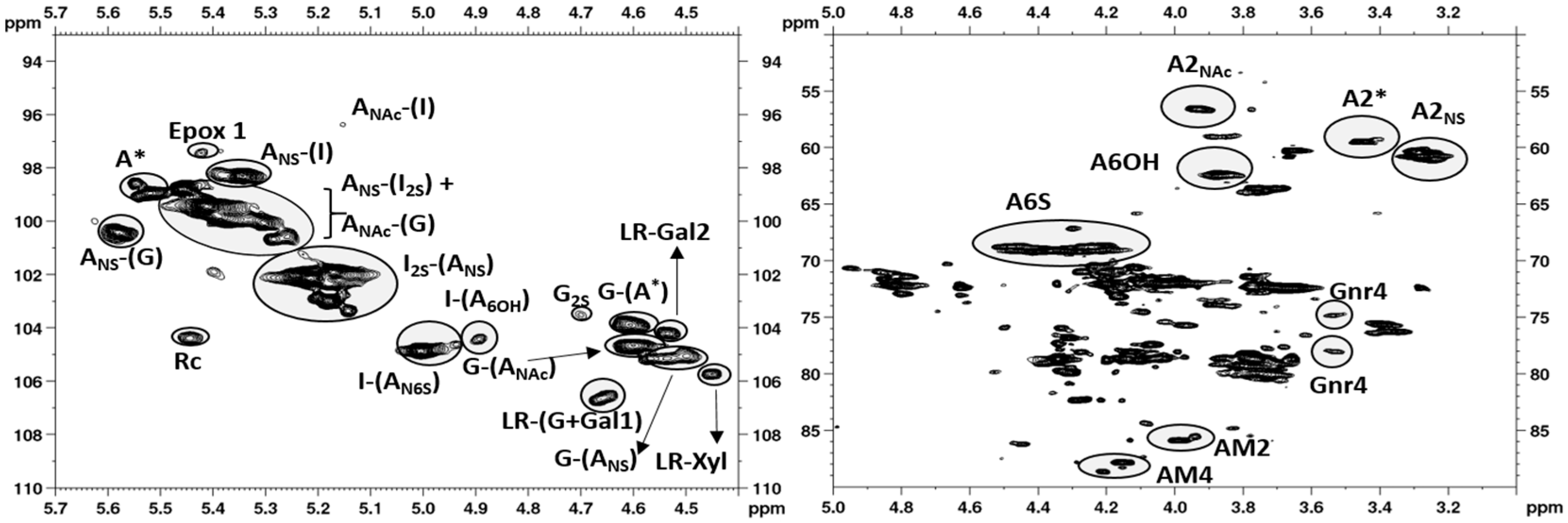
2.1. NMR Characterization
2.2. Chain Mapping
2.3. Building Blocks Analysis by Heparinases I, II, III Digestion
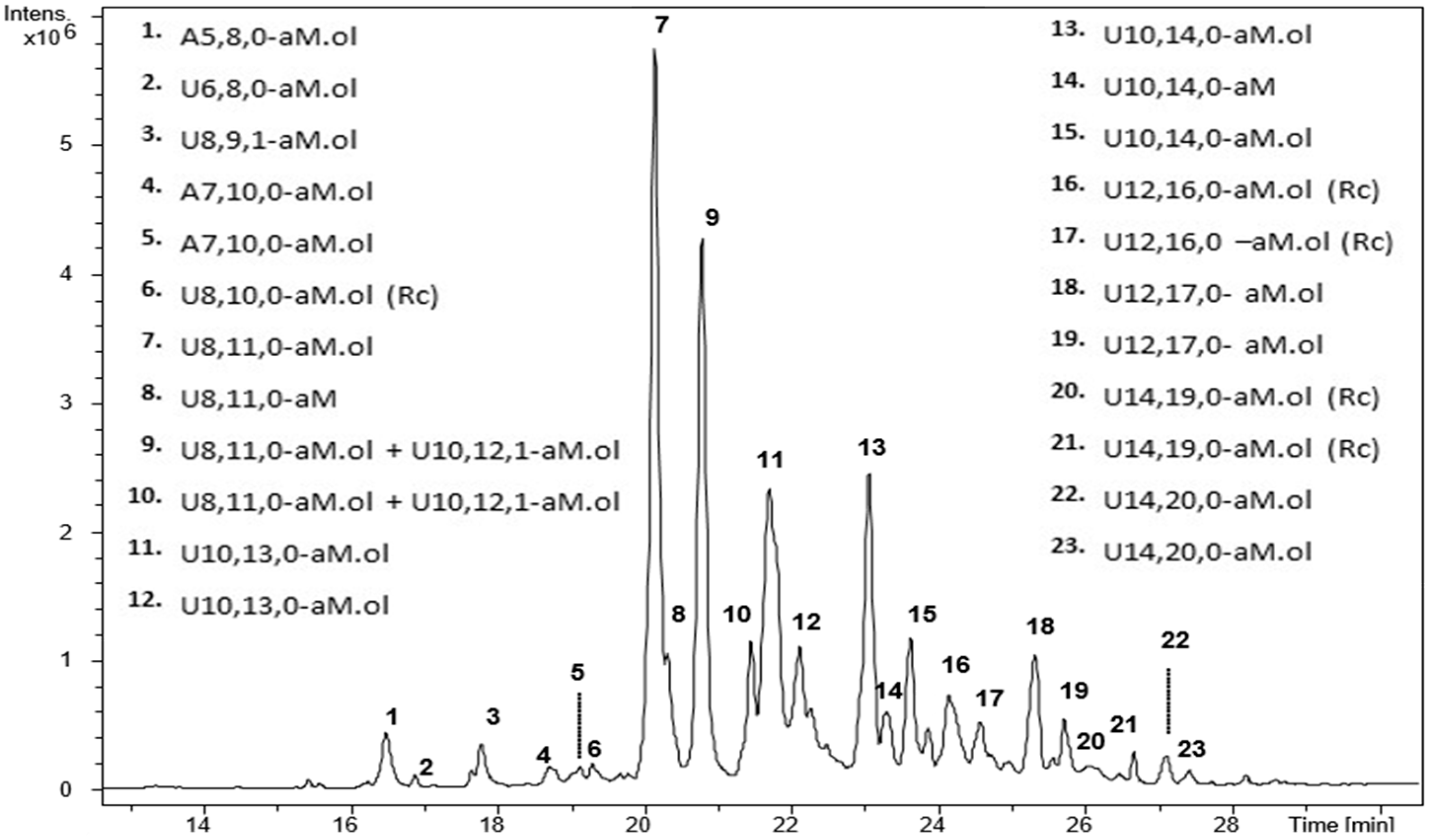
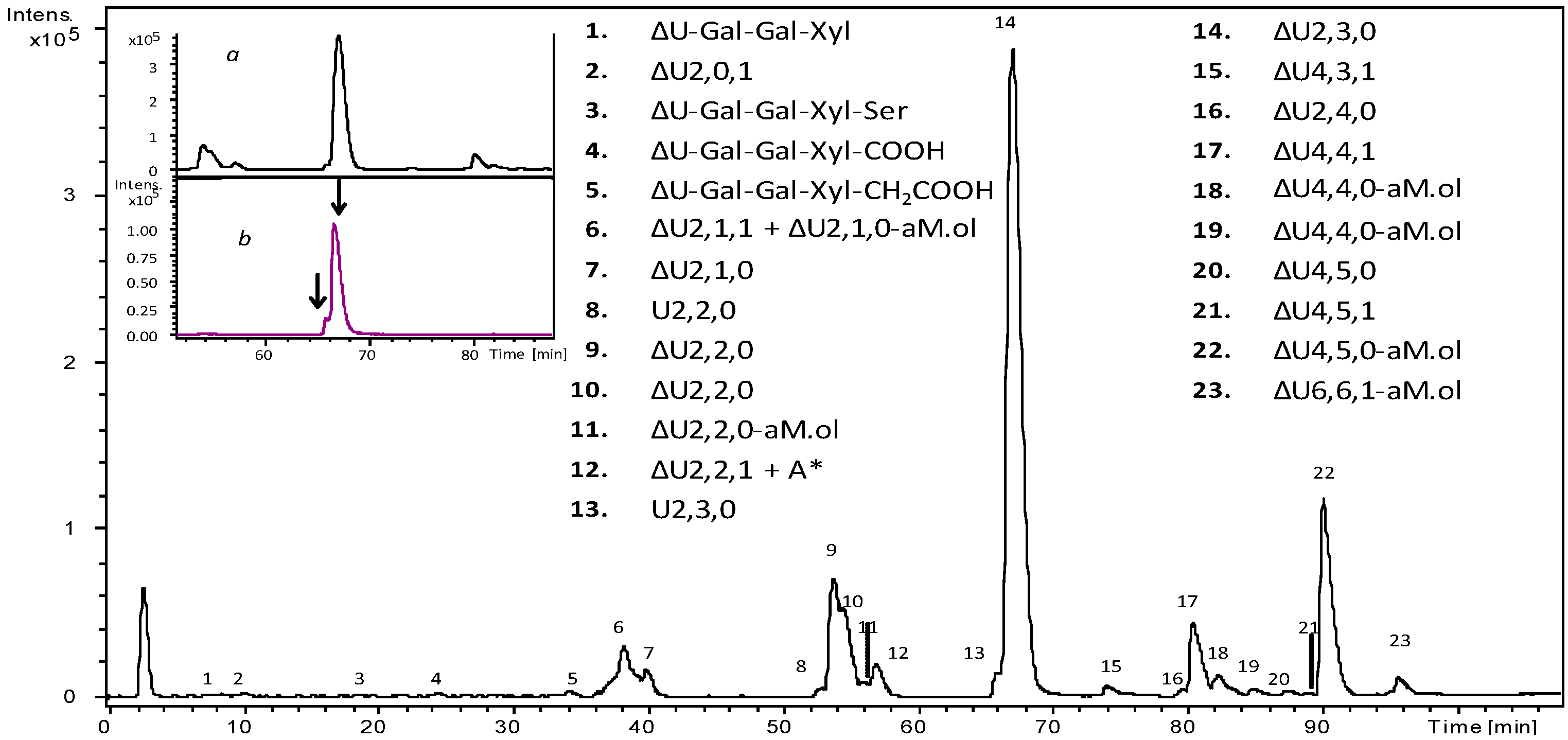
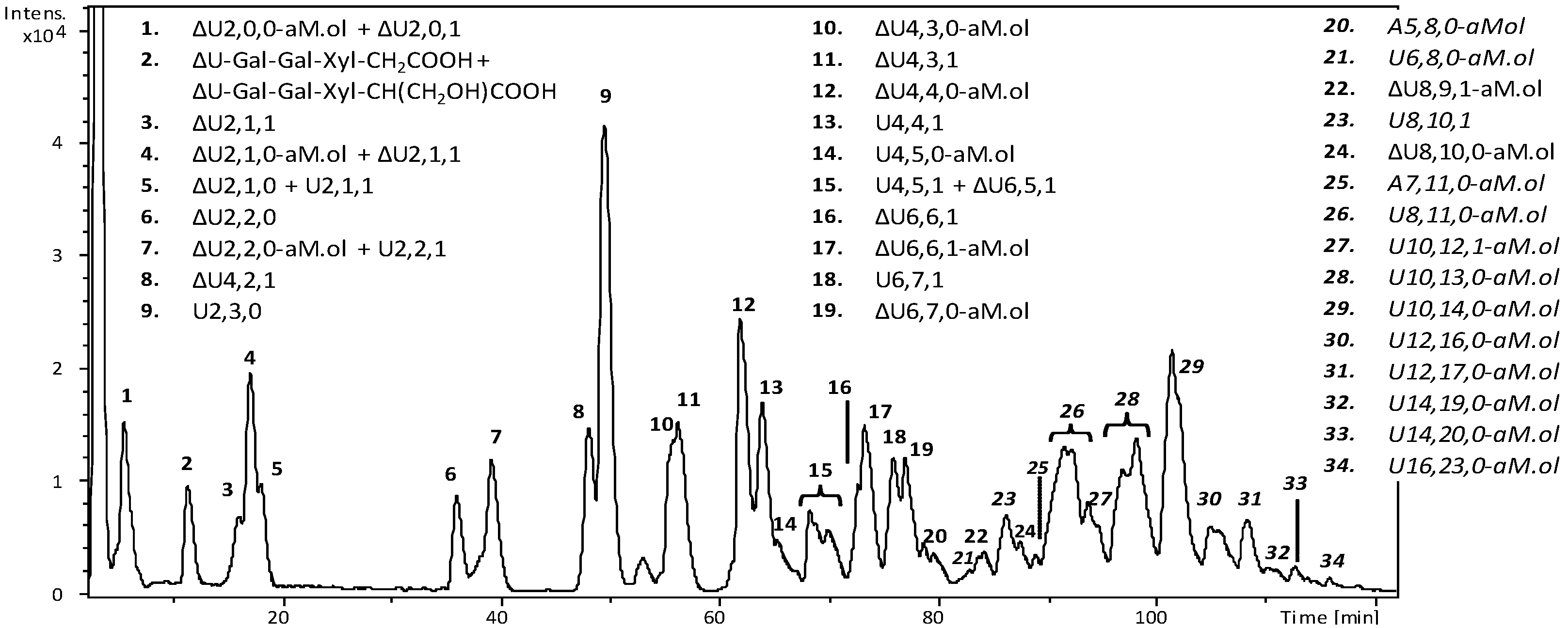
2.4. Bottom up Analysis by Heparinases III Digestion
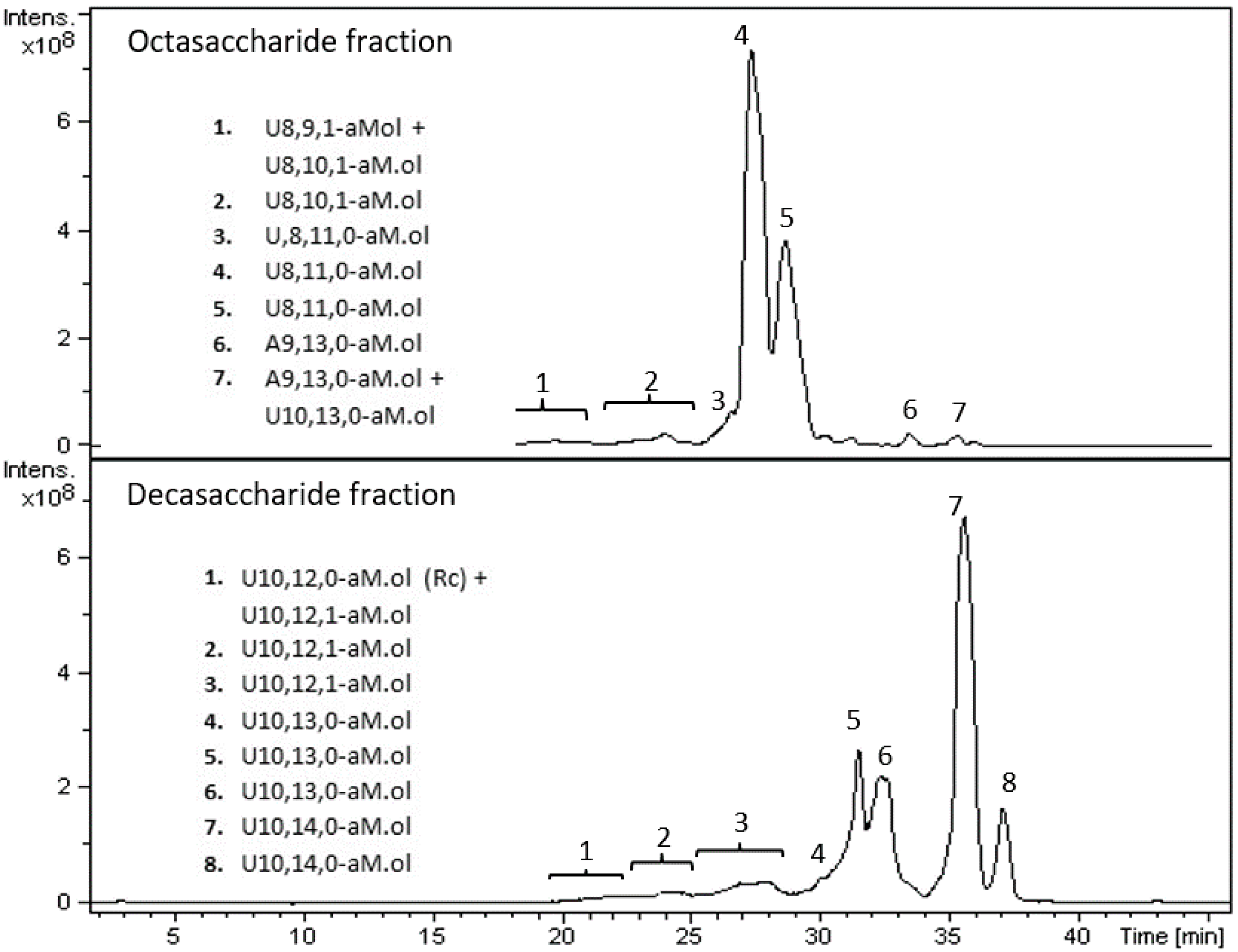
2.5. Isolation of Octasaccharide and Decasaccharide Fractions
2.6. Affinity Chromatography Separation of NA and HA Components
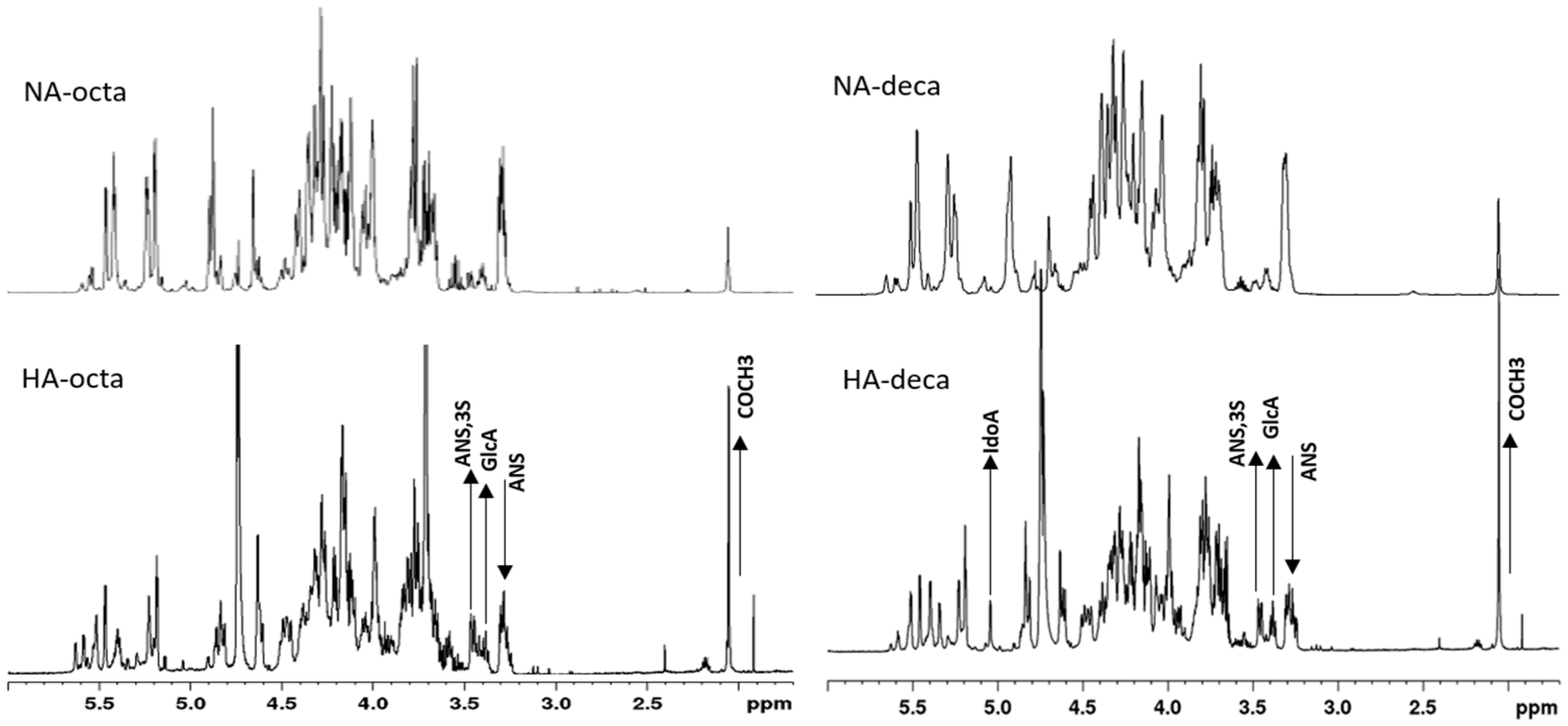
2.7. NMR Characterization of Octasaccharide and Decasaccharide Fractions and of their NA and HA Components
2.8. LC-MS Analysis of Octasaccharide and Decasaccharide fractions
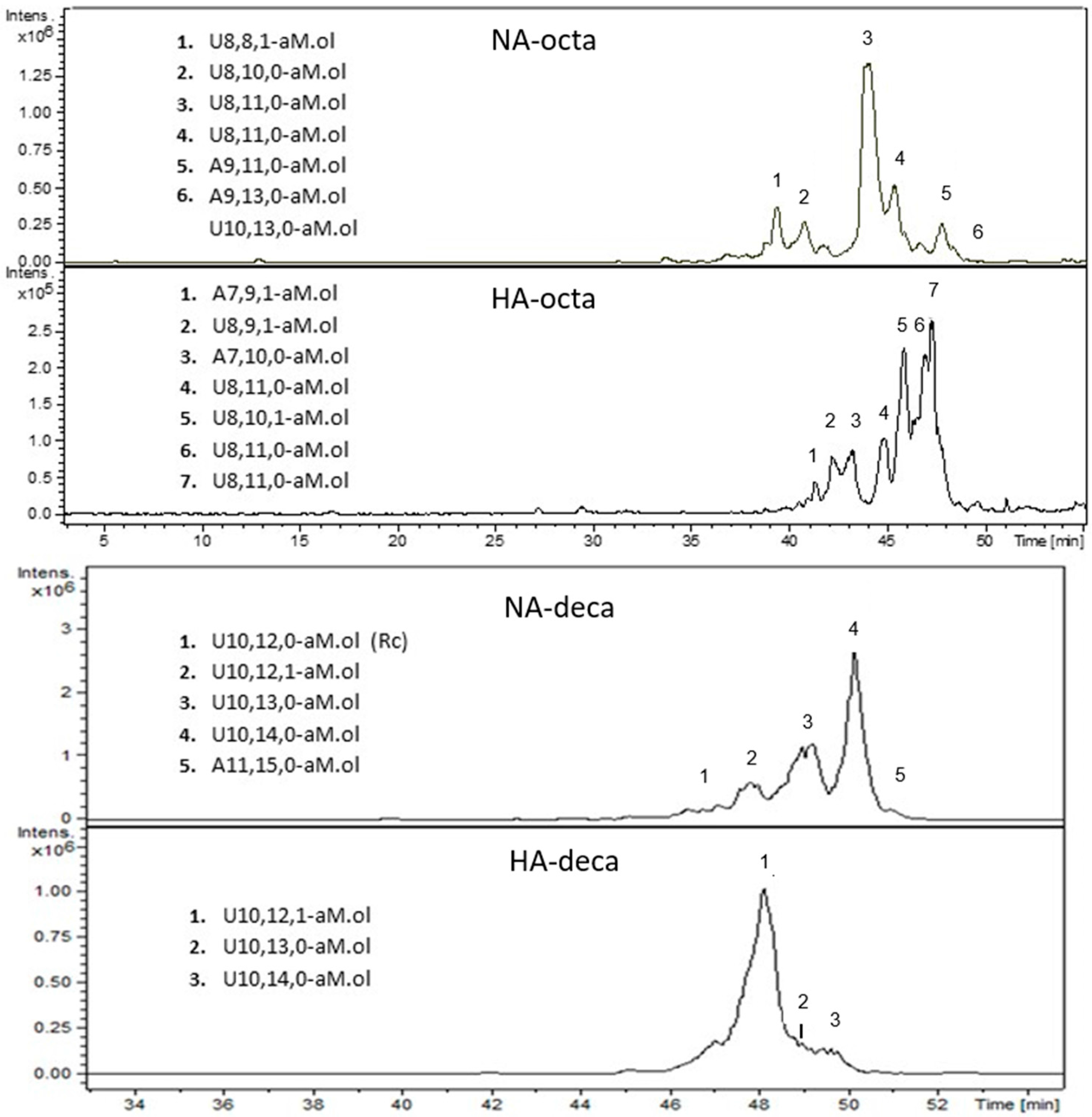
2.9. LC-MS Analysis of NA and HA components of Octasaccharide and Decasaccharide fractions
3. Materials and Methods
3.1. Materials
3.2. Fractionation by Size-Exclusion Chromatography
3.3. Desalting of Oligosaccharide Fractions
3.4. Affinity Chromatography on AT-Sepharose
3.5. NMR
3.6. Exhaustive enzymatic Depolymerization with Heparin Lyases
3.7. LC-MS Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fareed, J.; Jeske, W.; Hoppensteadt, D.; Clarizio, R.; Walenga, J.M. Low-molecular-weight heparins: Pharmacologic profile and product differentiation. Am. J. Cardiol. 1998, 82, 3L–10L. [Google Scholar] [CrossRef]
- Bisio, A.; Mantegazza, A.; Vecchietti, D.; Bensi, D.; Coppa, A.; Torri, G.; Bertini, S. Determination of the molecular weight of low-molecular-weight heparins by using high-pressure size exclusion chromatography on line with a triple detector array and conventional methods. Molecules 2015, 20, 5085–5098. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Bisio, A. Low-molecular-weight heparins: Differential characterization/physical characterization. In Heparin-A Century of Progress; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: London, UK, 2012; pp. 127–157. [Google Scholar]
- Hirsh, J.; Bauer, K.A.; Donati, M.B.; Samama, M.M.; Weitz, J.L. Parenteral anticoagulants: American college of chest physicians evidence-based clinical practice guidelines (8th edition). Chest 2008, 133 (Suppl. 6), 141S–159S. [Google Scholar] [CrossRef] [PubMed]
- Fareed, J.; Hoppensteadt, D.; Schultz, C.; Ma, Q.; Kujawski, M.F.; Neville, B.; Messmore, H. Biochemical and pharmacologic heterogeneity in low molecular weight heparins. Impact on the therapeutic profile. Curr. Pharm. Des. 2004, 10, 983–999. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Guglieri, S.; Naggi, A.; Sasisekharan, R.; Torri, G. Low molecular weight heparins: Structural differentiation by bidimensional nuclear magnetic resonance spectroscopy. Semin. Thromb. Hemost. 2007, 33, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Iacomini, M.; Casu, B.; Guerrini, M.; Naggi, A.; Pirola, A.; Torri, G. “Linkage region” sequence of heparin and heparan sulfate: Detection and quantification by nuclear magnetic resonance spectroscopy. Anal. Biochem. 1999, 274, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Fragmin (dalteparin sodium injection) Product Monograph, Pfizer Canada Inc, issued April 2016. Available online: www.pfizer.ca/sites/g/files/g10028126/f/201605/FRAGMIN_PM_193875_27April2016_E.pdf (accessed on 23 June 2017).
- Henricksen, J.; Ringborg, L.H.; Roepstorff, P. On-line size-exclusion chromatography/mass spectrometry of low molecular mass heparin. J. Mass Spectrom. 2004, 39, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Korir, A.; Larive, C.K. Advances in the separation, sensitive detection, and characterization of heparin and heparan sulfate. Anal. Bioanal. Chem. 2009, 393, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Zaia, J. On-line separations combined with MS for analysis of glycosaminoglycans. Mass Spectrom. Rev. 2008, 28, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Langeslay, D.J.; Urso, E.; Gardini, C.; Naggi, A.; Torri, G.; Larive, C.K. Reversed-phase ion-pair ultra-high-performance-liquid chromatography-mass spectrometry for fingerprinting low-molecular-weight heparins. J. Chromatogr. A 2013, 1292, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, A.; Casu, B.; Torri, G.; Pierro, S.; Naggi, A. Profiling glycol-split heparins by high-performance liquid chromatography/mass spectrometry analysis of their heparinase-generated oligosaccharides. Anal. Biochem. 2013, 434, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, X.; Zhu, Z.; Zhan, X.; Wu, Y.; Song, L.; Kang, J. Structural analysis of low molecular weight heparin by ultraperformance size exclusion chromatography/time of flight mass spectrometry and capillary zone electrophoresis. Anal. Chem. 2012, 85, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Buhse, L.F.; Al-Hakim, A.; Boyne li, M.T.; Keire, D.A. Characterization of currently marketed heparin products: Analysis of heparin digests by RPIP-UHPLC-QTOF-MS. J. Pharm. Biomed. Anal. 2012, 67–68, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, F.; Volpi, N. Novel reverse-phase ion pair-high performance liquid chromatography separation of heparin, heparan sulfate and low molecular weight-heparins disaccharides and oligosaccharides. J. Chromatogr. A 2013, 1284, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Wu, C.; Sun, X.; Liu, J.; Linhardt, R.J.; Zhang, Z. Development of hydrophilic interaction chromatography with quadruple time-of-flight mass spectrometry for heparin and low molecular weight heparin disaccharide analysis. Rapid Commun. Mass Spectrom. 2016, 30, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, Z.; Yu, M.; Lin, C.; Sheng, A.; Wang, Z.; Linhardt, R.J.; Chi, L. Hydrophilic interaction chromatography-multiple reaction monitoring mass spectrometry method for basic building block analysis of low-molecular-weight heparins prepared through nitrous acid depolymerization. J. Chromatogr. A 2017, 1479, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Mauri, L.; Boccardi, G.; Torri, G.; Karfunkle, M.; Macchi, E.; Muzi, L.; Keire, D.; Guerrini, M. Qualification of HSQC methods for quantitative composition of heparin and low molecular weight heparins. J. Pharm. Biomed. Anal. 2017, 136, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bisio, A.; Vecchietti, D.; Citterio, L.; Guerrini, M.; Raman, R.; Bertini, S.; Eisele, G.; Naggi, A.; Sasisekharan, R.; Torri, G. Structural features of low-molecular-weight heparins affecting their affinity to antithrombin. Thromb. Haemost. 2009, 102, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, A.; Casu, B.; Cassinelli, G.; Guerrini, M.; Torri, G.; Naggi, A. Structural features of glycol-split low-molecular-weight heparins and their heparin lyase generated fragments. Anal. Bioanal. Chem. 2014, 406, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Yoshida, K.; Sugiura, M.; Sugahara, K.; Khoo, K-H.; Morris, H.R.; Dell, A. Structural studies on the bacterial lyase-resistant tetrasaccharides derived from antithrombin III-binding site of porcine intestinal heparin. J. Biol. Chem. 1993, 268, 7, 4780–4787. [Google Scholar]
- Loganathan, D.; Wang, H.M.; Mallis, L.M.; Linhardt, R.J. Structural variation in the antithrombin III binding region and its occurrence in heparin from different sources. Biochemistry 1990, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hsieh, P.-H.; Xu, Y.; Thieker, D.; En Chai, E.J.; Xie, S.; Cooley, B.; Woods, R.; Chi, L.; Liu, J. Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc. 2017, 139, 5249–5256. [Google Scholar] [CrossRef] [PubMed]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Frag-1 | Frag-2 | Frag-3 | Frag-4 | Frag-5 | Frag-6 | Frag-7 | Frag-8 | Frag-9 | Frag-10 | Frag-11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Amines | |||||||||||
| ANS-(I2S) | 55.7 | 56.6 | 55.2 | 55.8 | 57.2 | 58.1 | 56.3 | 57.2 | 59.0 | 55.2 | 55.7 |
| ANS-I | 7.6 | 5.9 | 7.0 | 7.6 | 6.6 | 6.8 | 6.9 | 7.1 | 6.5 | 7.5 | 8.1 |
| ANS-(G) | 5.9 | 6.0 | 5.8 | 5.9 | 5.9 | 5.6 | 6.0 | 5.9 | 5.7 | 5.6 | 5.6 |
| A* | 5.9 | 5.5 | 6.1 | 5.3 | 5.6 | 4.9 | 5.5 | 5.2 | 4.7 | 5.5 | 5.1 |
| ANAc-(G) | 9.4 | 9.5 | 9.0 | 9.5 | 9.3 | 9.2 | 9.2 | 9.3 | 8.9 | 9.7 | 9.7 |
| ANAc-(I) | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| ANH2 | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| A-epox | nd | nd | 0.8 * | 0.6 * | 0.6 * | 0.7 * | 0.8 * | 0.6 * | nd | 1.1 * | nd |
| A-(GalA) | 1.6 | 1.9 | 1.4 * | 1.0 * | 1.1 * | 1.1 * | 0.5 * | 1.0 * | 0.9 * | 2.0 | 1.4 |
| A6S | 90.7 | 90.5 | 90.6 | 90.4 | 89.9 | 90.3 | 90.6 | 89.8 | 90.2 | 91.0 | 90.8 |
| aM.ol | 12.9 | 13.3 | 13.5 | 13.3 | 12.9 | 12.6 | 13.9 | 12.7 | 13.2 | 12.7 | 13.3 |
| Rc | 1.0 * | 1.2 * | 1.0 * | 0.9 * | 0.8 * | 1.0 * | 1.0 * | 0.9 * | 1.1 * | 0.7 * | 1.2 * |
| Uronic acids | |||||||||||
| I2S | 75.3 | 77.3 | 76.4 | 76.9 | 76.2 | 76.5 | 75.8 | 76.6 | 76.7 | 73.2 | 74.1 |
| I-(A6S) | 8.3 | 7.2 | 7.4 | 7.2 | 7.0 | 7.4 | 7.1 | 7.0 | 7.0 | 8.3 | 8.6 |
| I-(A6OH) | 0.6 | 0.7 * | 0.8 * | 1.0 * | 0.7 * | 0.7 * | 0.9 * | 0.9 * | 0.7 * | 0.8 * | 0.9 * |
| G-(A*) | 4.9 | 4.4 | 4.4 | 4.4 | 4.4 | 4.5 | 4.7 | 4.4 | 4.5 | 5.0 | 4.7 |
| G-(ANS) | 6.0 | 5.2 | 5.8 | 5.8 | 5.9 | 5.4 | 5.8 | 5.3 | 5.7 | 5.7 | 6.8 |
| G-(ANAc) | 3.9 | 4.6 | 3.9 | 4.1 | 4.5 | 4.7 | 4.9 | 4.5 | 5.3 | 4.1 | 3.8 |
| Gnr | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| epox | nd | nd | 0.8 * | 0.6 * | 0.6 * | 0.7 * | 0.7 * | 0.6 * | nd | 1.0 * | nd |
| GalA | 1.1 | 0.7 * | 0.5* | nd | 0.5 * | nd | nd | 0.7 * | nd | 1.7 | 1.1 * |
| Linkage region | |||||||||||
| Gal1+G | 1.6 * | 1.2 * | 1.6 | 2.1 | 2.0 | 1.8 | 2.1 | 2.0 | 1.9 | 1.3* | 1.4 |
| Gal2 | 1.3 * | 1.0 * | 1.3 * | 1.6 | 1.5 | 1.4 * | 1.6 | 1.4 | 1.6 | 1.2 * | 1.0 * |
| Xyl-Ser-ox | 0.8 * | 0.6 * | 0.8 * | 0.9 * | 0.9 * | 1.0 * | 1.0 * | 0.9 * | 1.1 | 0.6 * | 0.7 * |
| Xyl-Ser | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| Octa | HA-Octa | NA-Octa | Deca | HA-Deca | NA-Deca | |
|---|---|---|---|---|---|---|
| Amines | ||||||
| ANS-(I2S) | 56.6 | 34.5 | 58.6 | 58.1 | 27.0 | 59.8 |
| ANS-I | 2.8 | 4.1 | 2.3 | 4.6 | 13.8 | 4.1 |
| ANS-(G) | 4.4 | 10.3 | 2.8 | 5.3 | 9.0 | 5.1 |
| A* | 10.5 | 21.9 | 8.1 | 6.4 | 20.3 | 5.0 |
| ANAc-(G) | 2.2 | 7.2 | 2.0 | 4.4 | 11.6 | 3.3 |
| ANAc-(I) | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| ANH2 | n.d. | n.d. | 0.8 | n.d. | n.d. | n.d. |
| A-epox | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| A-(GalA) | 0.9 | n.d. | 0.8 | 0.9 | n.d. | n.d. |
| A6S | 98.5 | 99.5 | 98.8 | 97.0 | 99.5 | 97.4 |
| aM.ol | 20.8 | 21.6 | 22.6 | 19.0 | 18.2 | 18.7 |
| Rc | 1.0 | n.d. | 1.0 | 1.3 | n.d. | 1.5 |
| Uronic acids | ||||||
| I2S | 84.7 | 69.7 | 88.2 | 81.6 | 66.0 | 81.8 |
| I-(A6S) | 2.7 | 3.2 | 2.5 | 4.7 | 14.2 | 3.9 |
| I-(A6OH) | n.d. | n.d. | n.d. | 0.7 | n.d. | 0.6 |
| G-(A*) | 8.2 | 22.6 | 6.1 | 5.8 | 17.5 | 5.0 |
| G-(ANS) | 2.9 | 4.4 | 2.4 | 4.8 | 2.3 | 5.1 |
| G-(ANAc) | 0.6 | n.d. | n.d. | 1.5 | n.d. | 2.1 |
| Gnr | 4.6 | traces | 4.2 | 1.5 | traces | 2.9 |
| G2S | n.d. | traces | n.d. | n.d. | n.d. | n.d. |
| epox | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| GalA | 0.9 | n.d. | 0.7 | 0.9 | n.d. | 1.4 |
| Peak | Composition | Saccharide Sequence |
|---|---|---|
| 1 | A7,9,1-aM.ol | ANAc,6S-G-A*-I2S-ANS,6S-I2S-aM.ol6S |
| 2 | U8,9,1-aM.ol | I-ANAc,6S-G-A*-I2S-ANS,6S-I2S-aM.ol6S and/or I2S-ANS,6S-I-ANAc,6S-G-A*-I2S-aM.ol6S |
| 3 | A7,10,0-aM.ol | ANS,6S-G-A*-I2S-ANS,6S-I2S-aM.ol6S |
| 5 | U8,10,1-aM.ol | I2S-ANS,6S-I-ANAc,6S-I2S-A*-I2S-aM.ol6S |
| 4, 6, 7 | U8,11,0-aM.ol | I2S-ANS,6S-G-A*-I2S-ANS,6S-I2S-aM.ol6S I2S-ANS,6S-I2S-ANS,6S-G-A*-I2S-aM.ol6S G-A*-I2S-ANS,6S-I2S-ANS,6S-I2S-aM.ol6S |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisio, A.; Urso, E.; Guerrini, M.; De Wit, P.; Torri, G.; Naggi, A. Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies. Molecules 2017, 22, 1051. https://doi.org/10.3390/molecules22071051
Bisio A, Urso E, Guerrini M, De Wit P, Torri G, Naggi A. Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies. Molecules. 2017; 22(7):1051. https://doi.org/10.3390/molecules22071051
Chicago/Turabian StyleBisio, Antonella, Elena Urso, Marco Guerrini, Pauline De Wit, Giangiacomo Torri, and Annamaria Naggi. 2017. "Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies" Molecules 22, no. 7: 1051. https://doi.org/10.3390/molecules22071051
APA StyleBisio, A., Urso, E., Guerrini, M., De Wit, P., Torri, G., & Naggi, A. (2017). Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies. Molecules, 22(7), 1051. https://doi.org/10.3390/molecules22071051