The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2

 ,
,
Abstract
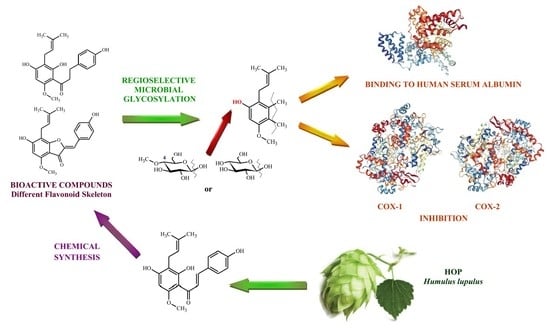
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
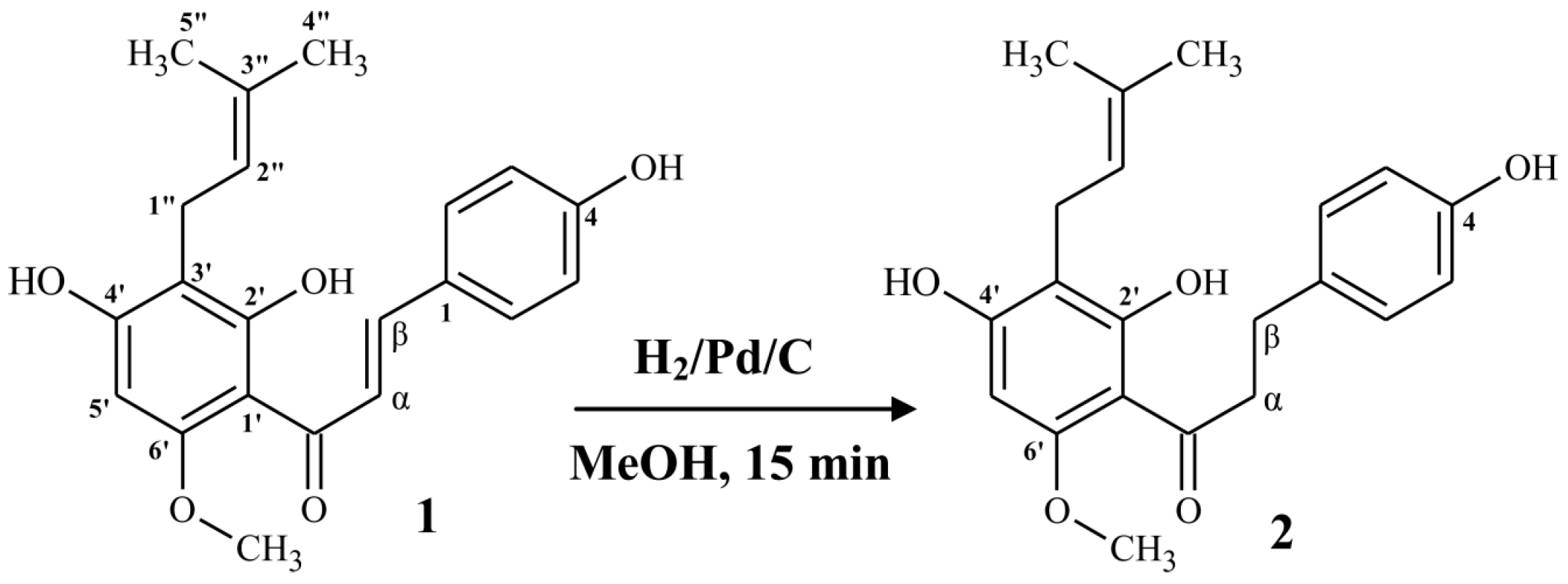
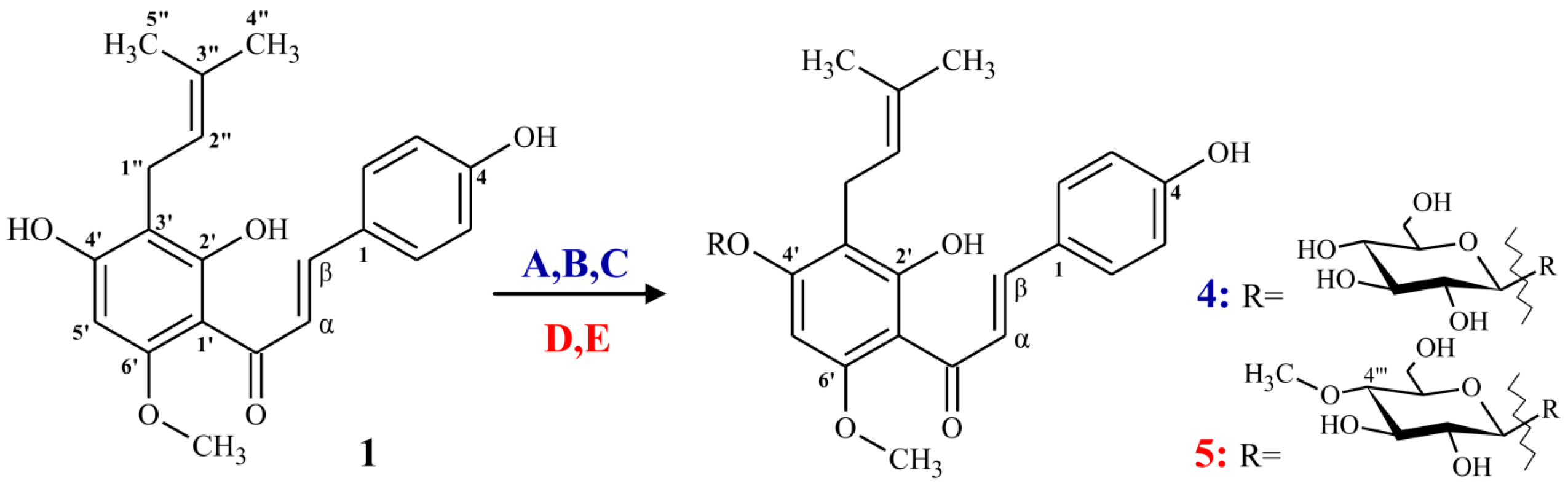
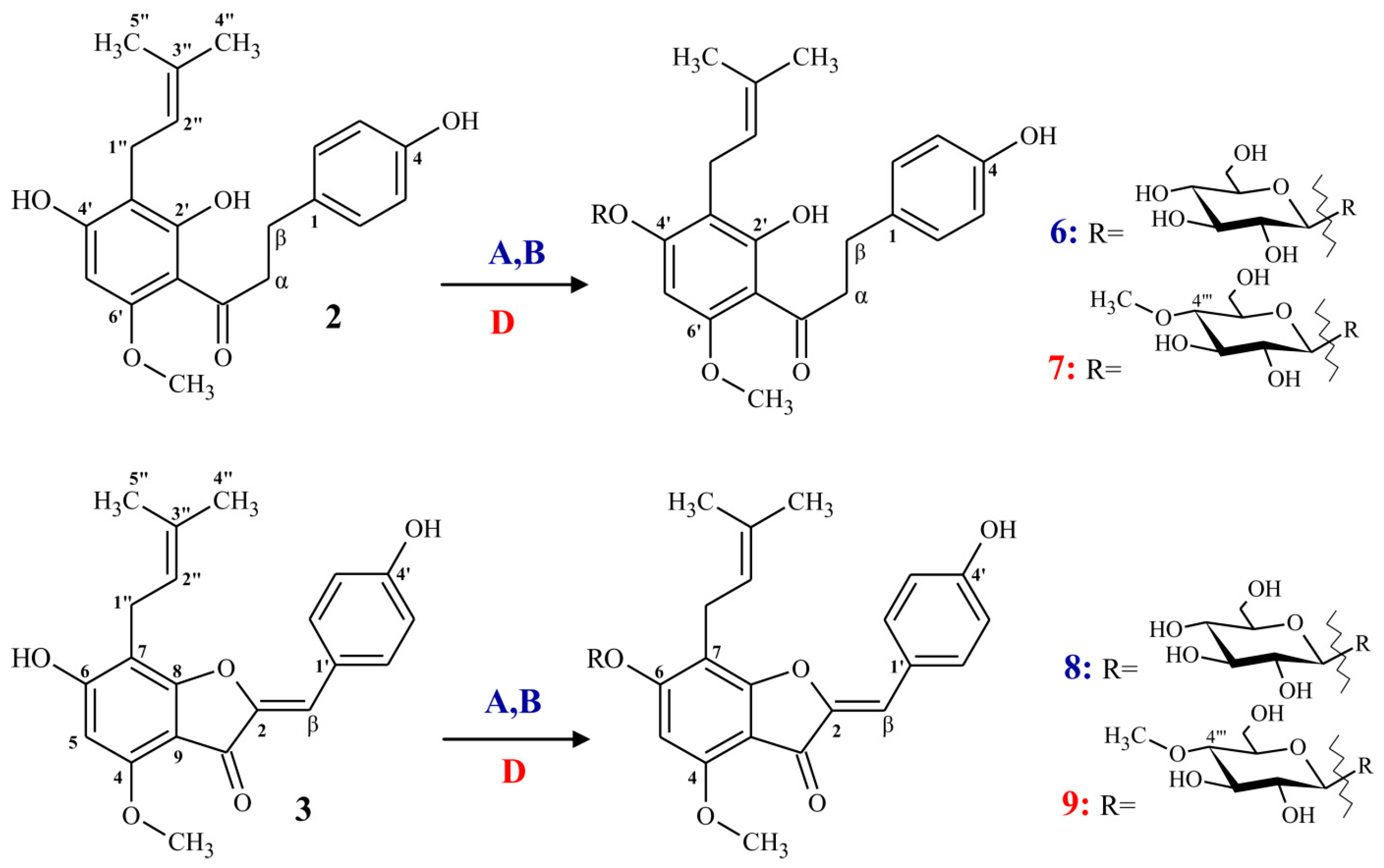
2.1.1. Synthesis of α,β-Dihydrochalcone (2)
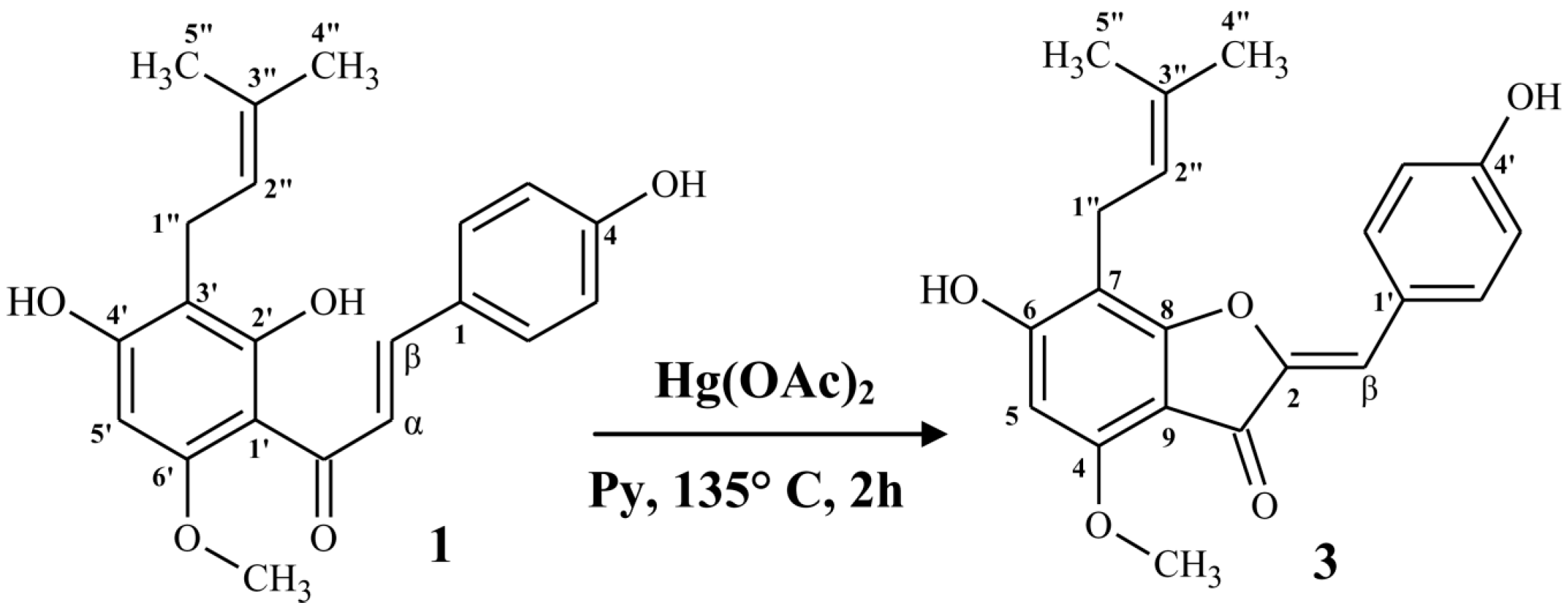
2.1.2. Synthesis of (Z)-6,4’-Dihydroxy-4-methoxy-7-prenylaurone (3)
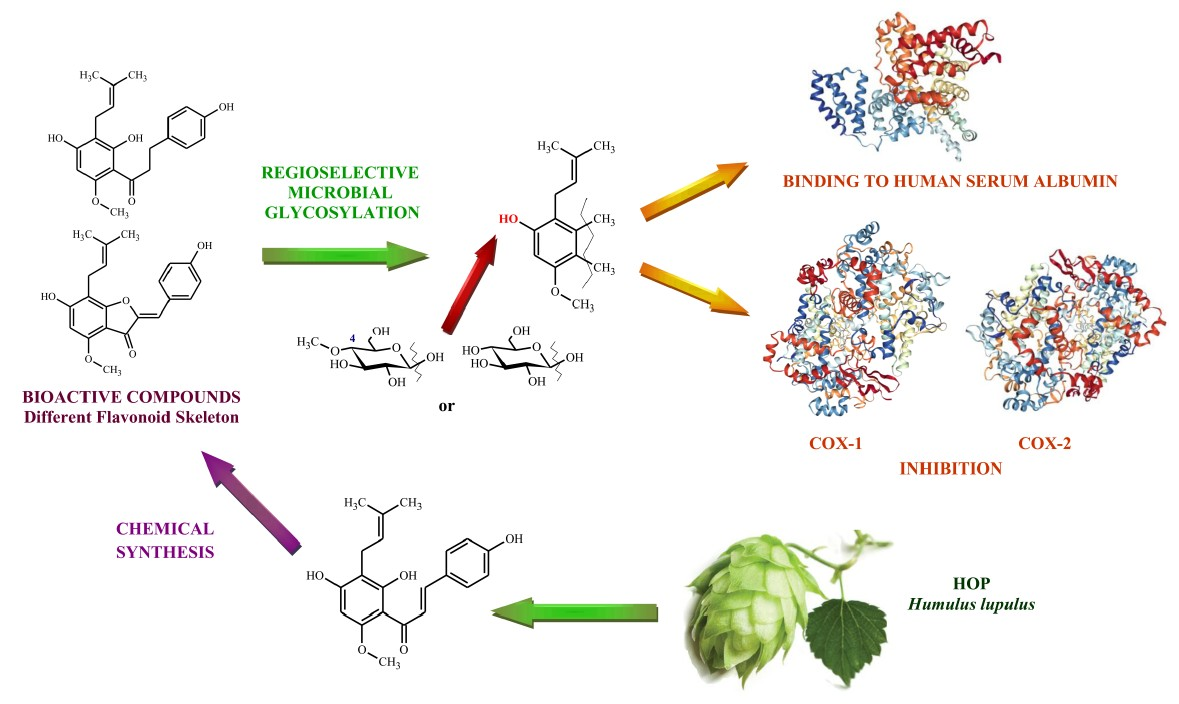
2.2. Regioselective Microbial Glycosylation of Prenylated Flavonoids
2.2.1. Biotransformations Catalyzed by Absidia coeruela AM93 and A. glauca AM177
2.2.2. Biotransformations Catalyzed by Beauveria bassiana AM278
2.3. Binding to Human Serum Albumin
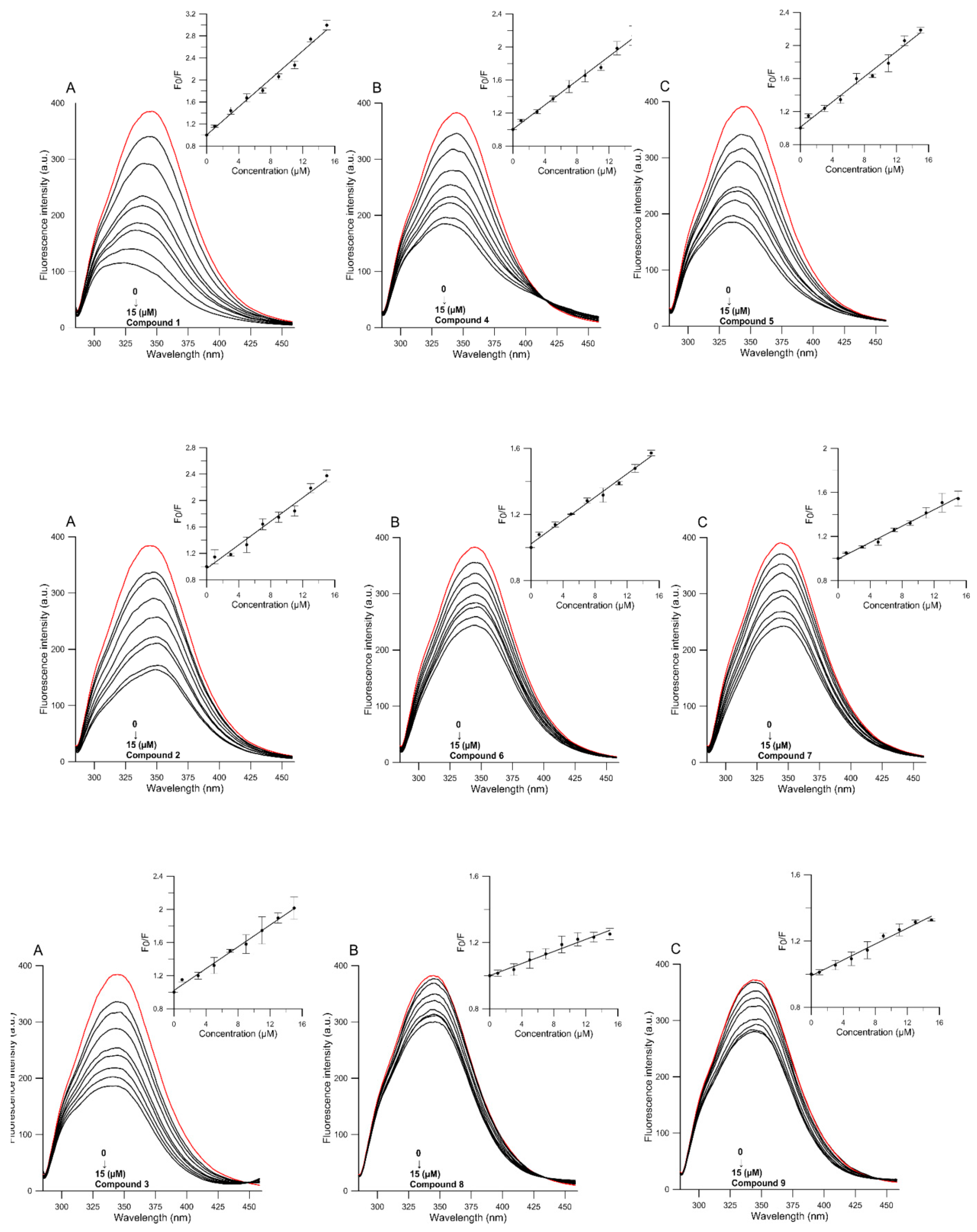
2.3.1. Fluorescence Quenching Mechanism of HSA
2.3.2. Binding Constant Kb and Number of Binding Sites
2.3.3. Thermodynamic Parameters Measurements
- (a)
- ∆H > 0 and ∆S > 0, hydrophobic forces;
- (b)
- ∆H < 0 and ∆S < 0, van der Waals interactions and hydrogen bonds; and
- (c)
2.4. Inhibition of Cyclooxygenases (COX-1 and COX-2) Activity
3. Material and Methods
3.1. General Experimental
3.2. Materials
3.2.1. Xanthohumol (1)
3.2.2. α,β-Dihydroxanthohumol (2)
3.2.3. (Z)-6,4’-Dihydroxy-4-methoxy-7-prenylaurone (3)
3.3. Biotransformation Products
3.4. Microorganisms
3.5. Conditions for Biotransformations
3.6. Products Isolation
3.7. Methods
3.7.1. Fluorescence Quenching of Human Serum Albumin
3.7.2. Cyclooxygenase (COX-1 and COX-2) Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stevens, J.F.; Page, J.E. Xanthohumol and related prenylflavonoids from hops and beer: To your good health! Phytochemistry 2004, 65, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Gerhäuser, C. Broad spectrum antiinfective potential of xanthohumol from hop (Humulus lupulus L.) in comparison with activities of other hop constituents and xanthohumol metabolites. Mol. Nutr. Food Res. 2005, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Hansen, P.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef] [PubMed]
- Tronina, T.; Bartmańska, A.; Filip-Psurska, B.; Wietrzyk, J.; Popłoński, J.; Huszcza, E. Fungal metabolites of xanthohumol with potent antiproliferative activity on human cancer cell lines in vitro. Bioorg. Med. Chem. 2013, 21, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, S.H.; Rupasinghe, H. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed]
- Tronina, T.; Bartmańska, A.; Popłoński, J.; Huszcza, E. Transformation of xanthohumol by Aspergillus ochraceus. J. Basic Microbiol. 2014, 54, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Bartmańska, A.; Huszcza, E.; Tronina, T. Transformation of isoxanthohumol by fungi. J. Mol. Catal. B Enzym. 2009, 61, 221–224. [Google Scholar] [CrossRef]
- Bartmańska, A.; Tronina, T.; Huszcza, E. Biotransformation of the phytoestrogen 8-prenylnaringenin. Z. Naturforsch. C 2010, 65, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Popłoński, J.; Sordon, S.; Tronina, T.; Bartmańska, A.; Huszcza, E. Fungal metabolism of naphthoflavones. J. Mol. Catal. B Enzym. 2015, 117, 1–6. [Google Scholar] [CrossRef]
- Sordon, S.; Madej, A.; Popłoński, J.; Bartmańska, A.; Tronina, T.; Brzezowska, E.; Juszczyk, P.; Huszcza, E. Regioselective ortho-hydroxylations of flavonoids by yeast. J. Agric. Food Chem. 2016, 64, 5525–5530. [Google Scholar] [CrossRef] [PubMed]
- Bartmańska, A.; Tronina, T.; Poploński, J.; Huszcza, E. Biotransformations of prenylated hop flavonoids for drug discovery and production. Curr. Drug Metab. 2013, 14, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Tronina, T.; Bartmańska, A.; Milczarek, M.; Wietrzyk, J.; Popłoński, J.; Rój, E.; Huszcza, E. Antioxidant and antiproliferative activity of glycosides obtained by biotransformation of xanthohumol. Bioorg. Med. Chem. Lett. 2013, 23, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Bartmańska, A.; Tronina, T.; Huszcza, E. Transformation of 8-prenylnaringenin by Absidia coerulea and Beauveria bassiana. Bioorg. Med. Chem. Lett. 2012, 22, 6451–6453. [Google Scholar] [CrossRef] [PubMed]
- Bartmańska, A.; Tronina, T.; Huszcza, E. Microbial sulfation of 8-prenylnaringenin. Z. Naturforsch. C 2013, 68, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Microbial glycosylation of daidzein, genistein and biochanin A: Two new glucosides of biochanin A. Molecules 2017, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- Hollman, P.C.; Bijsman, M.N.; van Gameren, Y.; Cnossen, E.P.; de Vries, J.H.; Katan, M.B. The sugar moiety is a major determinant of the absorption of dietary flavonoid glycosides in man. Free Radic. Res. 1999, 31, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [PubMed]
- Steensma, A.; Faassen-Peters, M.A.; Noteborn, H.P.; Rietjens, I.M. Bioavailability of genistein and its glycoside genistin as measured in the portal vein of freely moving unanesthetized rats. J. Agric. Food Chem. 2006, 54, 8006–8012. [Google Scholar] [CrossRef] [PubMed]
- Morand, C.; Manach, C.; Crespy, V.; Remesy, C. Respective bioavailability of quercetin aglycone and its glycosides in a rat model. Biofactors 2000, 12, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, S. Bioavailability of phenolic compounds. Crit. Rev. Food Sci. Nutr. 2004, 44, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Hollman, P.; Katan, M. Absorption, metabolism and health effects of dietary flavonoids in man. Biomed. Pharmacother. 1997, 51, 305–310. [Google Scholar] [CrossRef]
- Axelson, M.; Kirk, D.; Farrant, R.; Cooley, G.; Lawson, A.; Setchell, K. The identification of the weak oestrogen equol [7-hydroxy-3-(4’-hydroxyphenyl) chroman] in human urine. Biochem. J. 1982, 201, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Sjövall, J.; Gustafsson, B.; Setchell, K. Soya—A dietary source of the non-steroidal oestrogen equol in man and animals. J. Endocrinol. 1984, 102, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Brown, N.M.; Zimmer-Nechemias, L.; Brashear, W.T.; Wolfe, B.E.; Kirschner, A.S.; Heubi, J.E. Evidence for lack of absorption of soy isoflavone glycosides in humans, supporting the crucial role of intestinal metabolism for bioavailability. Am. J. Clin. Nutr. 2002, 76, 447–453. [Google Scholar] [PubMed]
- Andlauer, W.; Kolb, J.; Stehle, P.; Fürst, P. Absorption and metabolism of genistein in isolated rat small intestine. J. Nutr. 2000, 130, 843–846. [Google Scholar] [PubMed]
- Andlauer, W.; Kolb, J.; Fürst, P. Isoflavones from tofu are absorbed and metabolized in the isolated rat small intestine. J. Nutr. 2000, 130, 3021–3027. [Google Scholar] [PubMed]
- Huszcza, E.; Bartmańska, A.; Tronina, T. Glycosylation of xanthohumol by fungi. Z. Naturforsch. C 2008, 63, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U.; Chuang, V.T.G.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Trynda-Lemiesz, L.; Wiglusz, K. Effects of glycation on meloxicam binding to human serum albumin. J. Mol. Struct. 2011, 995, 35–40. [Google Scholar] [CrossRef]
- Xiao, J.; Muzashvili, T.S.; Georgiev, M.I. Advances in the biotechnological glycosylation of valuable flavonoids. Biotechnol. Adv. 2014, 32, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, Y.; He, L.-L.; Liu, B.; Zhang, S.-Y.; Ye, X.; Jing, J.-J.; Zhang, J.-F.; Gao, M.; Wang, X. Spectroscopic investigation on the food components-drug interaction: The influence of flavonoids on the affinity of nifedipine to human serum albumin. Food Chem. Toxicol. 2015, 78, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Etteldorf, N.; Etteldorf, N.; Becker, H. New chalcones from hop Humulus lupulus L. Z. Naturforsch. C 1999, 54, 610–612. [Google Scholar] [CrossRef]
- Stompor, M.; Potaniec, B.; Szumny, A.; Zieliński, P.; Żołnierczyk, A.; Anioł, M. Mikrobiologiczna redukcja ksantohumolu i 4-metoksychalkonu. Przem. Chem. 2013, 92, 574–578. [Google Scholar]
- Popłoński, J.; Tronina, T.; Sordon, S.; Huszcza, E. Selektywne uwodornienie ksantohumolu do α,β-dihydroksantohumolu. Przem. Chem. 2014, 93, 1916–1918. [Google Scholar]
- Stevens, J.F.; Miranda, C.L.; Frei, B.; Buhler, D.R. Inhibition of peroxynitrite-mediated LDL oxidation by prenylated flavonoids: The α,β-unsaturated keto functionality of 2’-hydroxychalcones as a novel antioxidant pharmacophore. Chem. Res. Toxicol. 2003, 16, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Venkateswarlu, S.; Panchagnula, G.K.; Gottumukkala, A.L.; Subbaraju, G.V. Synthesis, structural revision, and biological activities of 4’-chloroaurone, a metabolite of marine brown alga Spatoglossum variabile. Tetrahedron 2007, 63, 6909–6914. [Google Scholar] [CrossRef]
- Shanker, N.; Dilek, O.; Mukherjee, K.; McGee, D.; Bane, S. Aurones: Small molecule visible range fluorescent probes suitable for biomacromolecules. J. Fluoresc. 2011, 21, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bansal, M. Carbon-13 NMR of flavonoids. In Studies in Organic Chemistry Series 39; Agrawal, P.K., Ed.; Elsevier: Amsterdam, The Netherlands, 1989; pp. 236–282. [Google Scholar]
- Holland, H.L.; Morris, T.A.; Nava, P.J.; Zabic, M. A new paradigm for biohydroxylation by Beauveria bassiana ATCC 7159. Tetrahedron 1999, 55, 7441–7460. [Google Scholar] [CrossRef]
- Zhan, J.; Gunatilaka, A.A.L. Microbial transformation of amino- and hydroxyanthraquinones by Beauveria bassiana ATCC 7159. J. Nat. Prod. 2006, 69, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Gunatilaka, A.A.L. Microbial metabolism of 1-aminoanthracene by Beauveria bassiana. Bioorg. Med. Chem. 2008, 16, 5085–5089. [Google Scholar] [CrossRef] [PubMed]
- Olivo, H.F.; Peeples, T.L.; Rı́os, M.A.-Y.; Velázquez, F.; Kim, J.-W.; Narang, S. Microbial C-hydroxylation and β-4-O-methylglucosidation of methyl-benzamide 7-azanorbornane ethers with Beauveria bassiana. J. Mol. Catal. B Enzym. 2003, 21, 97–105. [Google Scholar] [CrossRef]
- Zhan, J.; Gunatilaka, A.A.L. Selective 4’-O-methylglycosylation of the pentahydroxy-flavonoid quercetin by Beauveria bassiana ATCC 7159. Biocatal. Biotransform. 2006, 24, 396–399. [Google Scholar] [CrossRef]
- Yuan, W.; Wang, P.; Zhang, Z.; Li, S. Glycosylation of (–)-maackiain by Beauveria bassiana and Cunninghamella echinulata var. elegans. Biocatal. Biotransform. 2010, 28, 117–121. [Google Scholar] [CrossRef]
- Naso, L.; Martínez, V.R.; Lezama, L.; Salado, C.; Valcarcel, M.; Ferrer, E.G.; Williams, P.A.M. Antioxidant, anticancer activities and mechanistic studies of the flavone glycoside diosmin and its oxidovanadium(IV) complex. Interactions with bovine serum albumin. Bioorg. Med. Chem. 2016, 24, 4108–4119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, D.; Wang, G.; Lu, Y. Study of interaction between human serum albumin and three antioxidants: Ascorbic acid, α-tocopherol, and proanthocyanidins. Eur. J. Med. Chem. 2013, 70, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Plenum Press: New York, NY, USA, 2006. [Google Scholar]
- Trnková, L.; Boušová, I.; Staňková, V.; Dršata, J. Study on the interaction of catechins with human serum albumin using spectroscopic and electrophoretic techniques. J. Mol. Struct. 2011, 985, 243–250. [Google Scholar] [CrossRef]
- Diniz, A.; Escuder-Gilabert, L.; Lopes, N.P.; Villanueva-Camañas, R.M.; Sagrado, S.; Medina-Hernández, M.J. Characterization of interactions between polyphenolic compounds and human serum proteins by capillary electrophoresis. Anal. Bioanal. Chem. 2008, 391, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Poór, M.; Boda, G.; Needs, P.W.; Kroon, P.A.; Lemli, B.; Bencsik, T. Interaction of quercetin and its metabolites with warfarin: Displacement of warfarin from serum albumin and inhibition of CYP2C9 enzyme. Biomed. Pharmacother. 2017, 88, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Klotz, I.M. Physicochemical aspects of drug-protein interactions: A general perspective. Ann. N. Y. Acad. Sci. 1973, 226, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, R.; Chi, Z.; Teng, Y.; Qin, P. New insights into the behavior of bovine serum albumin adsorbed onto carbon nanotubes: Comprehensive spectroscopic studies. J. Phys. Chem. B 2010, 114, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Del Tacca, M.; Colucci, R.; Fornai, M.; Blandizzi, C. Efficacy and tolerability of meloxicam, a COX-2 preferential nonsteroidal anti-inflammatory drug: A review. Clin. Drug Investig. 2002, 22, 799–818. [Google Scholar] [CrossRef]
- Gerhäuser, C.; Alt, A.; Heiss, E.; Gamal-Eldeen, A.; Klimo, K.; Knauft, J.; Neumann, I.; Scherf, H.-R.; Frank, N.; Bartsch, H.; et al. Cancer chemopreventive activity of xanthohumol, a natural product derived from hop. Mol. Cancer Ther. 2002, 1, 959–969. [Google Scholar] [PubMed]
- Crespy, V.; Morand, C.; Besson, C.; Manach, C.; Démigné, C.; Rémésy, C. Comparison of the intestinal absorption of quercetin, phloretin and their glucosides in rats. J. Nutr. 2001, 131, 2109–2114. [Google Scholar] [PubMed]
- Crespy, V.; Aprikian, O.; Morand, C.; Besson, C.; Manach, C.; Demigné, C.; Rémésy, C. Bioavailability of phloretin and phloridzin in rats. J. Nutr. 2001, 131, 3227–3230. [Google Scholar] [PubMed]
- Stevens, J.F.; Taylor, A.W.; Nickerson, G.B.; Ivancic, M.; Henning, J.; Haunold, A.; Deinzer, M.L. Prenylflavonoid variation in Humulus lupulus: Distribution and taxonomic significance of xanthogalenol and 4’-O-methylxanthohumol. Phytochemistry 2000, 53, 759–775. [Google Scholar] [CrossRef]
- Strugała, P.; Dudra, A.; Gabrielska, J. Interaction between mimic lipid membranes and acylated and nonacylated cyanidin and its bioactivity. J. Agric. Food Chem. 2016, 64, 7414–7422. [Google Scholar] [CrossRef] [PubMed]
- Strugała, P.; Gładkowski, W.; Kucharska, A.Z.; Sokół-Łętowska, A.; Gabrielska, J. Antioxidant activity and anti-inflammatory effect of fruit extracts from blackcurrant, chokeberry, hawthorn, and rosehip, and their mixture with linseed oil on a model lipid membrane. Eur. J. Lipid Sci. Technol. 2016, 118, 461–474. [Google Scholar] [CrossRef]
- Bourassa, P.; Dubeau, S.; Maharvi, G.M.; Fauq, A.H.; Thomas, T.; Tajmir-Riahi, H. Binding of antitumor tamoxifen and its metabolites 4-hydroxytamoxifen and endoxifen to human serum albumin. Biochimie 2011, 93, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Wach, A.; Pyrzyńska, K.; Biesaga, M. Quercetin content in some food and herbal samples. Food Chem. 2007, 100, 699–704. [Google Scholar] [CrossRef]
- Chang, Q.; Zuo, Z.; Chow, M.S.; Ho, W.K. Difference in absorption of the two structurally similar flavonoid glycosides, hyperoside and isoquercitrin, in rats. Eur. J. Pharm. Biopharm. 2005, 59, 549–555. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–9 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | T(K) | Ksv (104 M−1) | Kq (1012 M−1 s−1) | Kb (104 M−1) | n | ∆G (kJ/M) | ∆H (kJ/M) | ∆S (J/M/K) |
|---|---|---|---|---|---|---|---|---|
| 1 | 300 | 12.224 | 24.448 | 8.624 | 0.971 | −12.310 | −21.601 | −31.239 |
| 305 | 12.087 | 24.174 | 5.462 | 0.935 | −12.010 | |||
| 310 | 11.958 | 23.916 | 3.776 | 0.903 | −11.794 | |||
| 315 | 11.759 | 23.518 | 3.300 | 0.893 | −11.833 | |||
| 4 | 300 | 7.488 | 14.976 | 3.271 | 0.931 | −11.240 | −12.536 | −4.583 |
| 305 | 7.180 | 14.360 | 2.207 | 0.899 | −11.014 | |||
| 310 | 6.897 | 13.794 | 2.082 | 0.898 | −11.125 | |||
| 315 | 6.326 | 12.652 | 1.776 | 0.906 | −11.111 | |||
| 5 | 300 | 8.256 | 16.512 | 0.455 | 0.748 | −9.090 | −54.307 | −149.822 |
| 305 | 8.069 | 16.138 | 0.364 | 0.733 | −9.025 | |||
| 310 | 7.905 | 15.810 | 0.114 | 0.614 | −7.858 | |||
| 315 | 7.582 | 15.164 | 0.043 | 0.515 | −6.894 | |||
| 2 | 300 | 8.806 | 17.612 | 3.635 | 0.928 | −11.375 | −12.315 | −3.027 |
| 305 | 8.733 | 17.466 | 3.463 | 0.925 | −11.511 | |||
| 310 | 8.652 | 17.304 | 2.286 | 0.890 | −11.235 | |||
| 315 | 8.441 | 16.882 | 2.247 | 0.881 | −11.396 | |||
| 6 | 300 | 4.062 | 8.124 | 0.448 | 0.808 | −9.097 | −21.593 | −42.054 |
| 305 | 4.034 | 8.068 | 0.245 | 0.755 | −8.564 | |||
| 310 | 3.746 | 7.492 | 0.222 | 0.751 | −8.593 | |||
| 315 | 3.575 | 7.150 | 0.159 | 0.727 | −8.360 | |||
| 7 | 300 | 3.618 | 7.236 | 0.611 | 0.908 | −10.204 | −10.501 | −1.551 |
| 305 | 3.474 | 6.948 | 0.566 | 0.860 | −9.728 | |||
| 310 | 3.400 | 6.800 | 0.529 | 0.879 | −10.114 | |||
| 315 | 3.099 | 6.198 | 0.478 | 0.868 | −10.041 | |||
| 3 | 300 | 9.356 | 18.712 | 1.425 | 0.838 | −10.325 | −9.933 | −1.766 |
| 305 | 9.255 | 18.510 | 1.364 | 0.832 | −10.445 | |||
| 310 | 9.133 | 18.266 | 1.189 | 0.821 | −10.439 | |||
| 315 | 8.907 | 17.814 | 1.047 | 0.812 | −10.452 | |||
| 8 | 300 | 1.796 | 3.592 | 0.485 | 0.929 | −10.429 | −43.061 | −37.029 |
| 305 | 1.751 | 3.502 | 0.302 | 0.881 | −10.317 | |||
| 310 | 1.692 | 3.384 | 0.266 | 0.879 | −10.493 | |||
| 315 | 1.611 | 3.222 | 0.224 | 0.802 | −9.861 | |||
| 9 | 300 | 2.495 | 4.990 | 0.915 | 0.909 | −12.517 | −44.673 | −106.347 |
| 305 | 2.374 | 4.748 | 0.645 | 0.879 | −12.337 | |||
| 310 | 2.389 | 4.778 | 0.544 | 0.865 | −12.266 | |||
| 315 | 2.242 | 4.484 | 0.581 | 0.674 | −10.698 |
| Compound | IC50COX-1 (µM) | IC50COX-2 (µM) |
|---|---|---|
| 1 | 62.10 ± 3.48 | 51.86 ± 3.28 |
| 4 | 352.32 ± 10.91 | 302.95 ± 8.54 |
| 5 | 384.87 ±14.10 | 321.75 ±13.01 |
| 2 | 124.50 ± 7.61 | 103.8 ± 6.11 |
| 6 | 397.93 ± 7.51 | 370.81 ± 15.06 |
| 7 | 458.67 ± 4.02 | 451.18 ± 12.07 |
| 3 | 133.23 ± 6.91 | 109.02 ± 6.82 |
| 8 | 419.80 ± 8.34 | 405.01 ± 10.26 |
| 9 | 493.97 ± 6.34 | 482.16 ± 10.39 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tronina, T.; Strugała, P.; Popłoński, J.; Włoch, A.; Sordon, S.; Bartmańska, A.; Huszcza, E. The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2. Molecules 2017, 22, 1230. https://doi.org/10.3390/molecules22071230
Tronina T, Strugała P, Popłoński J, Włoch A, Sordon S, Bartmańska A, Huszcza E. The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2. Molecules. 2017; 22(7):1230. https://doi.org/10.3390/molecules22071230
Chicago/Turabian StyleTronina, Tomasz, Paulina Strugała, Jarosław Popłoński, Aleksandra Włoch, Sandra Sordon, Agnieszka Bartmańska, and Ewa Huszcza. 2017. "The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2" Molecules 22, no. 7: 1230. https://doi.org/10.3390/molecules22071230
APA StyleTronina, T., Strugała, P., Popłoński, J., Włoch, A., Sordon, S., Bartmańska, A., & Huszcza, E. (2017). The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2. Molecules, 22(7), 1230. https://doi.org/10.3390/molecules22071230