Structural Characterization of Cholestane Rhamnosides from Ornithogalum saundersiae Bulbs and Their Cytotoxic Activity against Cultured Tumor Cells


Abstract
:1. Introduction
2. Results and Discussion
2.1. Isolation and Structure Determination of 1–19
2.2. Cytotoxic Activity of 1–19, 1a, and 6a
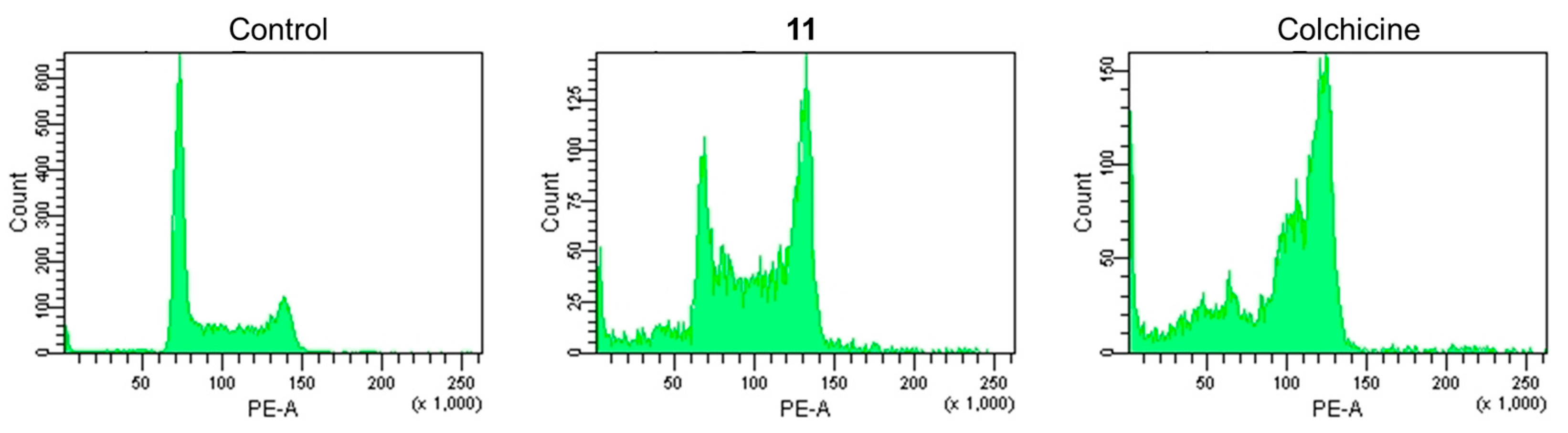
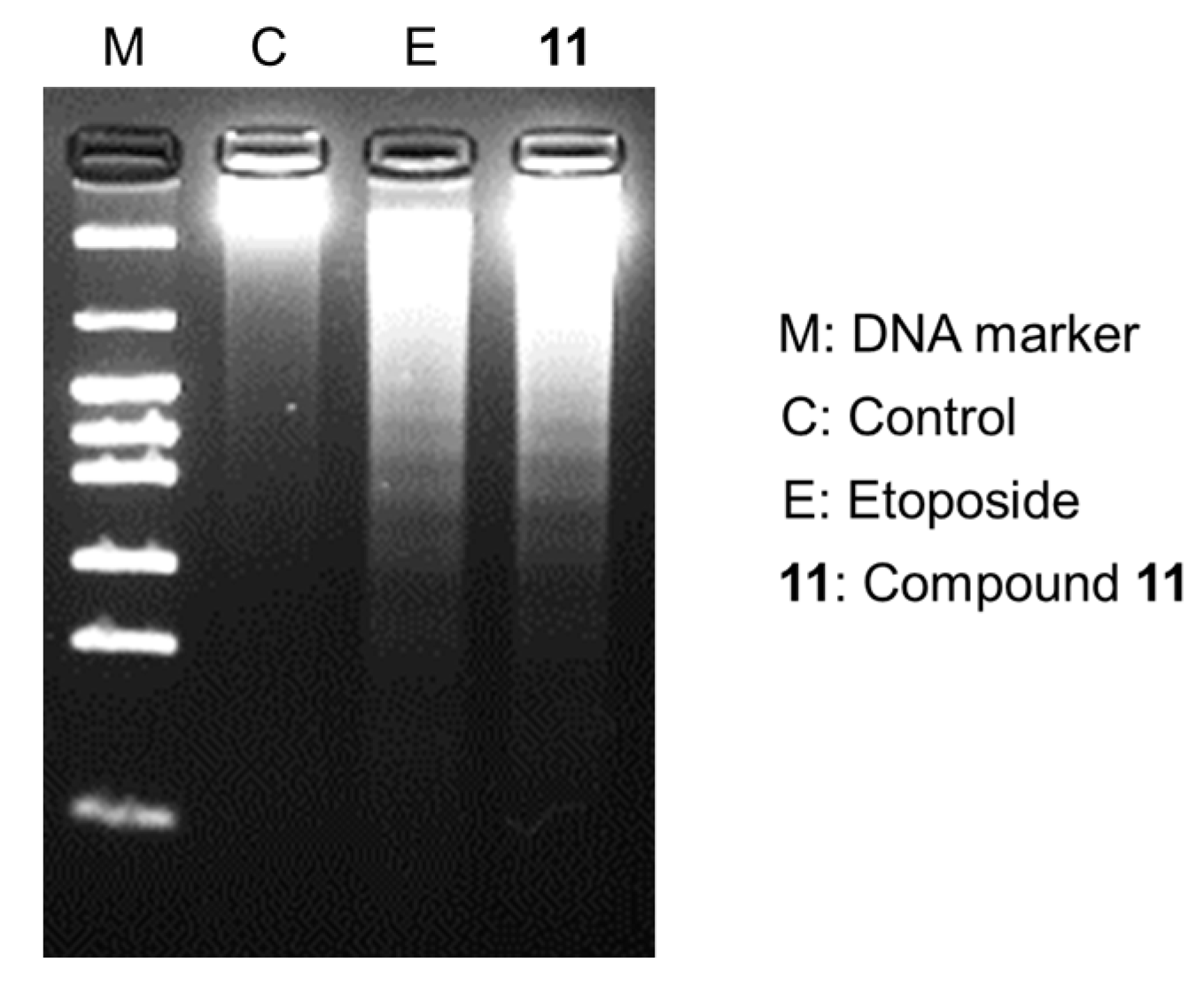
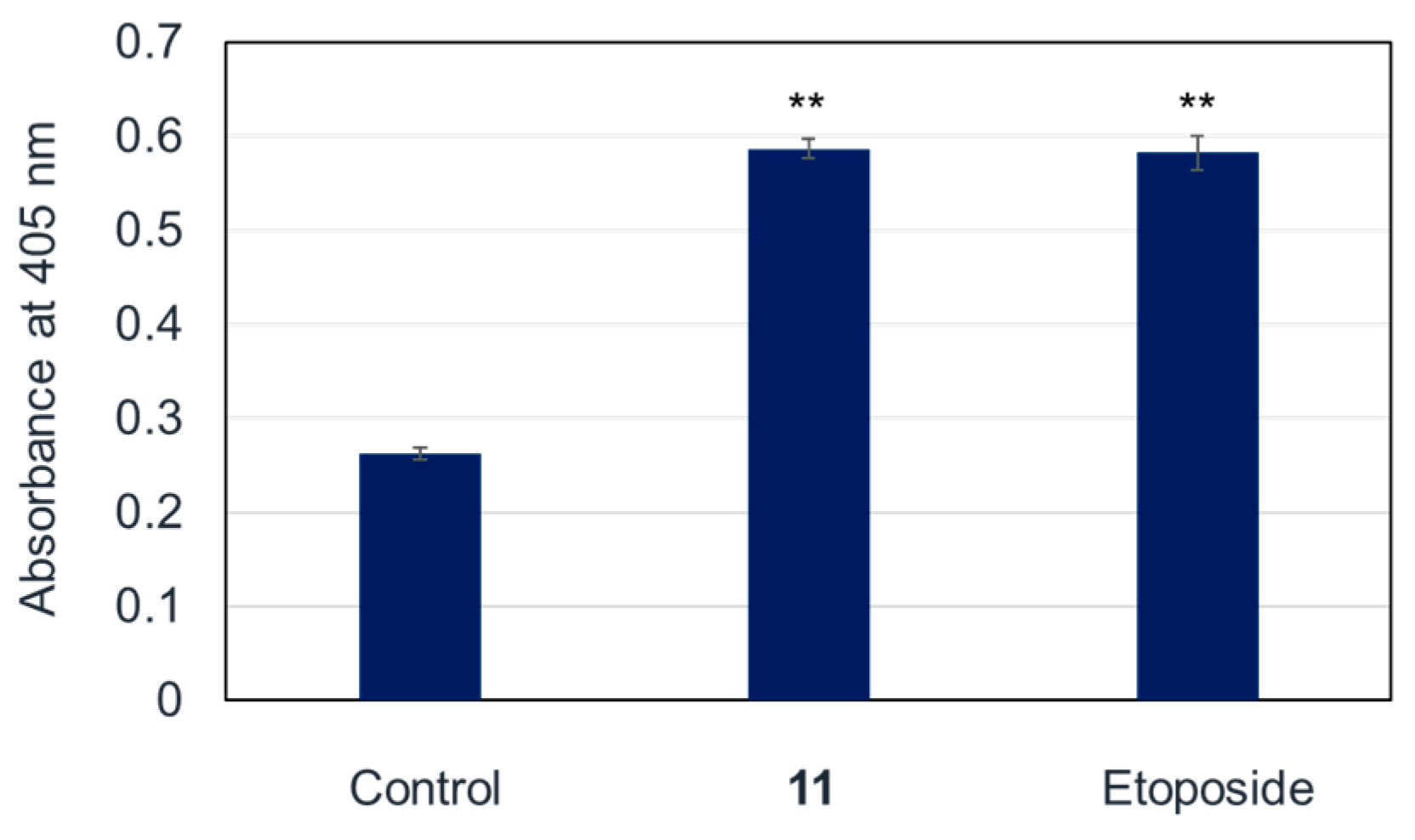
2.3. Apoptosis-Inducing Properties of 11 in HL-60 Cells
2.4. The Pathway of Apoptosis Induced by 11
3. Material and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Structural Characterization
3.5. Assay for Cytotoxic Activity
3.6. DAPI Staining
3.7. Cell Cycle Distribution Analysis
3.8. DNA Fragmentation Assay
3.9. Caspase-3 Activation Assay
3.10. Mitochondrial Membrane Potential (ΔΨm) Assay
3.11. Release of Cytochrome C into the Cytosol
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kuroda, M.; Mimaki, Y.; Sashida, Y.; Hirano, T.; Oka, K.; Dobashi, A. A novel 16, 23-epoxy-5β-cholestane glycoside with potent inhibitory activity on proliferation of human peripheral blood lymphocytes from Ornithogalum saundersiae bulbs. Chem. Pharm. Bull. 1995, 43, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Mimaki, Y.; Sashida, Y.; Nikaido, T.; Ohmoto, T. Structure of a novel 22-homo-23-norcholestane trisaccharide from Ornithogalum saundersiae. Tetrahedron Lett. 1993, 34, 6073–6076. [Google Scholar] [CrossRef]
- Mimaki, Y.; Kuroda, M.; Sashida, Y.; Hirano, T.; Oka, K.; Dobashi, A.; Koshino, H.; Uzawa, J. Three novel rearranged cholestane glycosides from Ornithogalum saundersiae bulbs and their cytostatic activities on leukemia HL-60 and MOlT-4 cells. Tetrahedron Lett. 1996, 37, 1245–1248. [Google Scholar] [CrossRef]
- Mimaki, Y.; Kuroda, M.; Kameyama, A.; Sashida, Y.; Hirano, T.; Oka, K.; Dobashi, A.; Koike, K.; Nikaido, T. A new rearranged cholestane glycoside from Ornithogalum saundersiae bulbs exhibiting potent cytostatic activities on leukemia HL-60 and MOLT-4 cells. Bioorg. Med. Chem. Lett. 1996, 6, 2635–2638. [Google Scholar] [CrossRef]
- Kuroda, M.; Mimaki, Y.; Sashida, Y.; Hirano, T.; Oka, K.; Dobashi, A.; Li, H.Y.; Harada, N. Novel cholestane glycosides from the bulbs of Ornithogalum saundersiae and their cytostatic activity on leukemia HL-60 and MOlT-4 cells. Tetrahedron 1997, 53, 11549–11562. [Google Scholar] [CrossRef]
- Kuroda, M.; Mimaki, Y.; Sashida, Y. Saundersiosides C–H, rearranged cholestane glycosides from the bulbs of Ornithogalum saundersiae and their cytostatic activity on HL-60 cells. Phytochemistry 1999, 52, 435–443. [Google Scholar] [CrossRef]
- Kuroda, M.; Mimaki, Y.; Sashida, Y. Cholestane rhamnosides from the bulbs of Ornithogalum saundersiae. Phytochemistry 1999, 52, 445–452. [Google Scholar] [CrossRef]
- Mimaki, Y.; Kuroda, M.; Kameyama, A.; Sashida, Y.; Hirano, T.; Oka, K.; Koike, K.; Nikaido, T. A new cytotoxic cholestane bisdesmoside from Ornithogalum saundersiae bulbs. Biosci. Biotechnol. Biochem. 1996, 60, 1049–1050. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Mimaki, Y.; Terao, M.; Nikaido, T.; Ohmoto, T. Acylated cholestane glycosides the bulbs of Ornithogalum saundersiae. Phytochemistry 1992, 31, 3969–3973. [Google Scholar] [CrossRef]
- Mimaki, Y.; Kuroda, M.; Kameyama, A.; Sashida, Y.; Hirano, T.; Oka, K.; Maekawa, R.; Wada, T.; Sugita, K.; Beutler, J.A. Cholestane glycosides with potent cytostatic activities on various tumor cells from Ornithogalum saundersiae bulbs. Bioorg. Med. Chem. Lett. 1997, 7, 633–636. [Google Scholar] [CrossRef]
- Kubo, S.; Mimaki, Y.; Sashida, Y.; Nikaido, T.; Ohmoto, T. New polyhydroxylated cholestane glycosides from the bulbs of Ornithogalum saundersiae. Chem. Pharm. Bull. 1992, 40, 2469–2472. [Google Scholar] [CrossRef]
- Kuroda, M.; Mimaki, Y.; Yokosuka, A.; Sashida, Y.; Beutler, J.A. Cytotoxic cholestane glycosides from the bulbs of Ornithogalum saundersiae. J. Nat. Prod. 2001, 64, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Mimaki, Y.; Kuroda, M.; Yokosuka, A.; Sashida, Y. Five New polyoxygenated cholestane bisdesmosides from the bulbs of Galtonia candicans. J. Nat. Prod. 2001, 64, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, Z.; Wu, H.; Yokosuka, A.; Mimaki, Y. Steroidal glycosides from the underground parts of Dracaena thalioides and their cytotoxic activity. Phytochemistry 2014, 107, 102–110. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 1a | 2 | 3 | ||||||||||||||||
| Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | ||||||||
| Glc 1 | 5.09 | d | 7.7 | Glc 1 | 5.27 | d | 8.1 | Glc 1 | 5.06 | d | 7.7 | Glc 1 | 5.08 | d | 7.1 | ||||
| 2 | 4.28 | dd | 9.0, 7.7 | 2 | 4.07 | dd | 8.1, 8.1 | 2 | 4.05 | dd | 8.9, 7.7 | 2 | 4.07 | dd | 8.8, 7.1 | ||||
| 3 | 4.29 | dd | 9.0, 9.0 | 3 | 4.26 | m | 3 | 4.29 | dd | 8.9, 8.8 | 3 | 4.33 | dd | 8.8, 8.8 | |||||
| 4 | 4.29 | dd | 9.0, 9.0 | 4 | 4.42 | m | 4 | 4.28 | dd | 8.8, 8.8 | 4 | 4.30 | dd | 8.8, 8.8 | |||||
| 5 | 3.99 | m | 5 | 3.99 | m | 5 | 3.96 | m | 5 | 3.31 | m | ||||||||
| 6 | a | 4.54 | dd | 11.8, 2.6 | 6 | a | 4.54 | dd | 11.9, 2.1 | 6 | a | 4.52 | dd | 11.8, 2.2 | 6 | a | 4.53 | dd | 11.8, 2.3 |
| b | 4.41 | dd | 11.8, 6.8 | b | 4.41 | dd | 11.9, 6.8 | b | 4.39 | dd | 11.8, 5.0 | b | 4.43 | dd | 11.8, 5.3 | ||||
| Rha 1 | 5.05 | br s | Rha 1 | 5.08 | br s | Rha 1 | 5.00 | br s | Rha 1 | 5.08 | d | 1.9 | |||||||
| 2 | 5.66 | br d | 2.6 | 2 | 4.48 | br s | 2 | 5.62 | br d | 3.3 | 2 | 5.78 | dd | 3.0, 1.9 | |||||
| 3 | 4.54 | dd | 9.3, 2.6 | 3 | 4.44 | dd | 8.6, 3.2 | 3 | 5.76 | dd | 10.0, 3.3 | 3 | 6.05 | dd | 9.4, 3.0 | ||||
| 4 | 4.19 | dd | 9.3, 9.3 | 4 | 4.29 | m | 4 | 4.20 | dd | 10.0, 10.0 | 4 | 4.38 | dd | 9.4, 9.4 | |||||
| 5 | 4.26 | m | 5 | 4.29 | m | 5 | 4.33 | dq | 10.0, 6.2 | 5 | 4.42 | m | |||||||
| 6 | 1.71 | d | 6.1 | 6 | 1.68 | d | 6.1 | 6 | 1.67 | d | 6.2 | 6 | 1.75 | d | 5.7 | ||||
| Ac | 2.03 | s | Rha 2-O-Ac | 2.07 | s | PMBz 1 | - | ||||||||||||
| Rha 3-O-Ac | 1.98 | s | 2 | 8.19 | d | 8.8 | |||||||||||||
| 3 | 6.93 | d | 8.8 | ||||||||||||||||
| 4 | - | ||||||||||||||||||
| 5 | 6.93 | d | 8.8 | ||||||||||||||||
| 6 | 8.19 | d | 8.8 | ||||||||||||||||
| 7 | - | ||||||||||||||||||
| OMe | 3.65 | s | |||||||||||||||||
| Ac | 2.12 | s | |||||||||||||||||
| 4 | 5 | 6 | |||||||||||||||||
| Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | |||||||||||
| Glc 1 | 5.08 | d | 8.1 | Rha 1 | 5.07 | br s | Glc 1 | 5.07 | d | 7.7 | |||||||||
| 2 | 4.07 | dd | 8.6, 8.1 | 2 | 5.65 | br d | 2.2 | 2 | 4.06 | dd | 8.8, 7.7 | ||||||||
| 3 | 4.32 | dd | 8.9, 8.6 | 3 | 4.55 | dd | 9.4, 2.2 | 3 | 4.30 | dd | 8.8, 8.8 | ||||||||
| 4 | 4.29 | dd | 8.9, 8.9 | 4 | 4.20 | dd | 9.4, 9.4 | 4 | 4.29 | dd | 8.8, 8.8 | ||||||||
| 5 | 3.98 | m | 5 | 4.29 | m | 5 | 3.97 | m | |||||||||||
| 6 | a | 4.54 | dd | 11.9, 2.4 | 6 | 1.71 | d | 6.5 | 6 | a | 4.53 | dd | 11.6, 2.0 | ||||||
| b | 4.42 | dd | 11.9, 6.8 | b | 4.41 | dd | 11.6, 4.9 | ||||||||||||
| Ac | 2.03 | s | |||||||||||||||||
| Rha 1 | 5.10 | d | 1.7 | Rha 1 | 5.03 | br s | |||||||||||||
| 2 | 5.81 | dd | 3.3, 1.7 | 2 | 5.63 | br d | 3.1 | ||||||||||||
| 3 | 6.08 | dd | 9.6, 3.3 | 3 | 5.78 | dd | 9.7, 3.1 | ||||||||||||
| 4 | 4.38 | dd | 9.6, 9.6 | 4 | 4.21 | dd | 9.7, 9.7 | ||||||||||||
| 5 | 4.46 | m | 5 | 4.34 | dq | 9.7, 6.1 | |||||||||||||
| 6 | 1.76 | d | 6.0 | 6 | 1.68 | d | 6.1 | ||||||||||||
| TMBz 1 | - | Rha 2-O-Ac | 2.09 | s | |||||||||||||||
| 2 | 7.54 | s | Rha 3-O-Ac | 1.98 | s | ||||||||||||||
| 3 | - | ||||||||||||||||||
| 4 | - | ||||||||||||||||||
| 5 | - | ||||||||||||||||||
| 6 | 7.54 | s | |||||||||||||||||
| 7 | - | ||||||||||||||||||
| TMBz 3, 5-OMe | 3.73 | s | |||||||||||||||||
| TMBz 4-OMe | 3.88 | s | |||||||||||||||||
| Ac | 2.17 | s | |||||||||||||||||
| 6a | 7 | 8 | 9 | ||||||||||||||||
| Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | ||||||||
| Glc 1 | 5.09 | dd | 7.7 | Glc 1 | 5.08 | d | 7.6 | Glc 1 | 5.09 | d | 7.6 | Glc 1 | 5.07 | d | 7.3 | ||||
| 2 | 4.07 | dd | 8.3, 7.7 | 2 | 4.07 | dd | 9.0, 7.6 | 2 | 4.07 | dd | 8.8, 7.6 | 2 | 4.28 | dd | 9.2, 7.3 | ||||
| 3 | 4.31 | m | 3 | 4.33 | dd | 9.0, 9.0 | 3 | 4.33 | dd | 8.8, 8.8 | 3 | 4.31 | m | ||||||
| 4 | 4.30 | m | 4 | 4.30 | dd | 9.0, 9.0 | 4 | 4.31 | dd | 8.8, 8.8 | 4 | 4.14 | m | ||||||
| 5 | 3.99 | m | 5 | 3.98 | m | 5 | 3.99 | m | 5 | 4.03 | m | ||||||||
| 6 | a | 4.55 | br d | 10.8 | 6 | a | 4.54 | dd | 11.9, 2.2 | 6 | a | 4.55 | dd | 11.6, 2.7 | 6 | a | 4.49 | dd | 11.9, 2.3 |
| b | 4.41 | m | b | 4.42 | m | b | 4.43 | dd | 11.6, 6.2 | b | 4.35 | dd | 11.9, 6.2 | ||||||
| Rha 1 | 5.24 | br s | Rha 1 | 5.10 | d | 1.4 | Rha 1 | 5.12 | d | 1.6 | Rha 1 | 6.39 | br s | ||||||
| 2 | 4.50 | br s | 2 | 5.78 | dd | 2.7, 1.4 | 2 | 5.81 | dd | 3.1, 1.6 | 2 | 4.82 | br d | 3.2 | |||||
| 3 | 4.43 | m | 3 | 6.08 | dd | 9.7, 2.7 | 3 | 6.11 | dd | 9.6, 3.1 | 3 | 4.67 | dd | 9.2, 3.2 | |||||
| 4 | 4.31 | m | 4 | 4.38 | dd | 9.7, 9.7 | 4 | 4.39 | dd | 9.6, 9.6 | 4 | 4.35 | m | ||||||
| 5 | 4.28 | m | 5 | 4.42 | m | 5 | 4.46 | m | 5 | 5.06 | m | ||||||||
| 6 | 1.68 | d | 5.6 | 6 | 1.75 | d | 5.9 | 6 | 1.76 | d | 6.0 | 6 | 1.75 | d | 6.0 | ||||
| PMBz 1 | - | TMBz 1 | - | Rha' 1 | 5.07 | br s | |||||||||||||
| 2 | 8.18 | d | 8.9 | 2 | 7.52 | s | 2 | 5.65 | br d | 3.1 | |||||||||
| 3 | 6.93 | d | 8.9 | 3 | - | 3 | 5.81 | dd | 9.9, 3.1 | ||||||||||
| 4 | - | 4 | - | 4 | 4.23 | m | |||||||||||||
| 5 | 6.93 | d | 8.9 | 5 | - | 5 | 4.37 | m | |||||||||||
| 6 | 8.18 | d | 8.9 | 6 | 7.52 | s | 6 | 1.75 | d | 6.0 | |||||||||
| 7 | - | 7 | - | ||||||||||||||||
| OMe | 3.65 | s | TMBz 3, 5-OMe | 3.70 | s | Rha' 2-O-Ac | 2.10 | s | |||||||||||
| TMBz 4-OMe | 3.88 | s | Rha' 3-O-Ac | 1.98 | s | ||||||||||||||
| Ac | 2.13 | s | |||||||||||||||||
| Ac | 2.19 | s | |||||||||||||||||
| 10 | 11 | 12 | |||||||||||||||||
| Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | |||||||||||
| Glc 1 | 5.08 | d | 7.7 | Rha 1 | 5.25 | br s | Glc 1 | 5.06 | d | 7.7 | |||||||||
| 2 | 4.08 | dd | 8.7, 7.7 | 2 | 4.52 | br d | 2.8 | 2 | 4.07 | dd | 8.7, 7.7 | ||||||||
| 3 | 4.31 | dd | 8.7, 8.7 | 3 | 4.45 | dd | 8.6, 2.8 | 3 | 4.33 | dd | 8.7. 8.7 | ||||||||
| 4 | 4.31 | dd | 8.7, 8.7 | 4 | 4.32 | dd | 8.6, 8.6 | 4 | 4.29 | dd | 8.7, 8.7 | ||||||||
| 5 | 4.00 | m | 5 | 4.30 | m | 5 | 4.00 | m | |||||||||||
| 6 | a | 4.59 | dd | 11.8, 2.3 | 6 | 1.67 | d | 5.7 | 6 | a | 4.57 | dd | 11.9, 2.3 | ||||||
| b | 4.44 | dd | 11.8, 5.3 | b | 4.42 | dd | 11.9, 5.3 | ||||||||||||
| Rha 1 | 5.03 | d | 1.7 | Rha 1 | 5.04 | br s | |||||||||||||
| 2 | 5.66 | dd | 3.3, 1.7 | 2 | 5.65 | br d | 3.0 | ||||||||||||
| 3 | 5.81 | dd | 9.7, 3.3 | 3 | 5.80 | dd | 9.7, 3.0 | ||||||||||||
| 4 | 4.22 | dd | 9.7, 9.7 | 4 | 4.22 | dd | 9.7, 9.7 | ||||||||||||
| 5 | 4.35 | m | 5 | 4.35 | m | ||||||||||||||
| 6 | 1.70 | d | 6.2 | 6 | 1.69 | d | 6.1 | ||||||||||||
| Rha 2-O-Ac | 2.10 | s | Rha 2-O-Ac | 2.09 | s | ||||||||||||||
| Rha 3-O-Ac | 2.00 | s | Rha 3-O-Ac | 1.98 | s | ||||||||||||||
| Positions | 1 | 1a | 2 | 3 | 4 | 5 | 6 | 6a | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 39.9 | 39.8 | 39.4 | 39.6 | 39.7 | 40.0 | 39.6 | 39.6 | 39.6 | 39.6 | 39.6 | 37.4 | 37.7 | 37.3 |
| 2 | 30.5 | 30.4 | 30.2 | 32.1 | 30.5 | 32.2 | 30.4 | 30.5 | 30.4 | 30.4 | 30.4 | 30.3 | 32.6 | 30.2 |
| 3 | 78.2 | 78.3 | 78.0 | 78.2 | 78.4 | 71.7 | 78.1 | 78.2 | 78.2 | 78.2 | 78.2 | 78.0 | 71.2 | 78.0 |
| 4 | 39.7 | 39.6 | 39.6 | 39.8 | 39.9 | 44.1 | 39.8 | 39.9 | 39.8 | 39.8 | 39.7 | 39.3 | 43.4 | 39.2 |
| 5 | 141.8 | 141.8 | 141.6 | 141.8 | 142.0 | 142.9 | 141.8 | 141.8 | 141.8 | 141.8 | 141.8 | 141.0 | 142.0 | 140.9 |
| 6 | 121.5 | 121.5 | 121.2 | 121.4 | 121.4 | 120.8 | 121.4 | 121.5 | 121.4 | 121.4 | 121.5 | 121.7 | 121.1 | 121.7 |
| 7 | 32.1 | 32.1 | 31.9 | 30.4 | 32.2 | 32.9 | 32.1 | 32.2 | 32.1 | 32.1 | 32.2 | 32.0 | 32.0 | 31.9 |
| 8 | 31.7 | 31.6 | 31.4 | 31.5 | 31.7 | 31.8 | 31.5 | 31.7 | 31.6 | 31.6 | 31.7 | 31.8 | 31.9 | 31.8 |
| 9 | 57.0 | 56.9 | 56.6 | 56.8 | 57.0 | 57.1 | 56.8 | 57.1 | 56.8 | 56.9 | 56.9 | 50.3 | 50.4 | 50.2 |
| 10 | 38.8 | 38.8 | 38.6 | 38.7 | 38.9 | 38.8 | 38.7 | 38.8 | 38.7 | 38.8 | 38.9 | 36.9 | 36.9 | 36.8 |
| 11 | 68.1 | 68.1 | 67.8 | 68.0 | 68.1 | 68.1 | 68.0 | 68.2 | 68.0 | 68.0 | 68.0 | 21.1 | 21.1 | 21.0 |
| 12 | 51.7 | 51.7 | 51.4 | 51.6 | 51.8 | 51.8 | 51.6 | 51.7 | 51.7 | 51.7 | 51.7 | 40.0 | 40.0 | 39.9 |
| 13 | 42.9 | 42.9 | 42.6 | 42.8 | 42.9 | 43.0 | 42.8 | 42.9 | 42.8 | 42.8 | 42.9 | 42.2 | 42.2 | 42.1 |
| 14 | 54.4 | 54.4 | 54.1 | 54.3 | 54.4 | 54.4 | 54.2 | 54.4 | 54.2 | 54.3 | 54.3 | 54.9 | 55.0 | 54.7 |
| 15 | 35.5 | 35.5 | 35.2 | 35.3 | 35.4 | 35.6 | 35.3 | 35.6 | 35.3 | 35.3 | 35.6 | 35.4 | 35.6 | 35.3 |
| 16 | 82.7 | 82.3 | 83.0 | 83.2 | 83.3 | 82.9 | 83.2 | 82.2 | 83.2 | 83.2 | 83.4 | 83.0 | 82.0 | 82.9 |
| 17 | 57.7 | 57.8 | 57.4 | 57.6 | 57.8 | 57.7 | 57.6 | 57.8 | 57.6 | 57.6 | 57.7 | 57.7 | 57.7 | 57.6 |
| 18 | 14.3 | 14.3 | 14.0 | 14.2 | 14.3 | 14.4 | 14.2 | 14.4 | 14.2 | 14.2 | 14.2 | 13.1 | 13.2 | 13.0 |
| 19 | 19.1 | 19 | 18.8 | 19.0 | 19.1 | 19.3 | 19.0 | 19.1 | 19.0 | 19.0 | 19.1 | 19.4 | 19.6 | 19.3 |
| 20 | 35.9 | 35.9 | 35.9 | 36.1 | 36.2 | 35.4 | 35.6 | 35.1 | 35.6 | 35.6 | 38.9 | 36.2 | 35.1 | 35.6 |
| 21 | 11.9 | 11.8 | 11.8 | 12.0 | 12.0 | 11.9 | 12.0 | 11.8 | 12.0 | 12.0 | 12.0 | 12.1 | 11.8 | 12.1 |
| 22 | 72.4 | 72.4 | 72.5 | 72.6 | 72.7 | 71.8 | 72.0 | 71.9 | 72.0 | 72.0 | 72.0 | 72.8 | 72.0 | 72.1 |
| 23 | 34.6 | 34.3 | 34.3 | 34.5 | 34.6 | 35.4 | 35.4 | 35.3 | 35.4 | 35.5 | 35.5 | 34.6 | 35.3 | 35.4 |
| 24 | 36.4 | 36.7 | 36.4 | 36.5 | 36.6 | 123.5 | 123.5 | 123.0 | 123.0 | 123.5 | 123.5 | 36.7 | 123.1 | 123.5 |
| 25 | 29.0 | 28.6 | 28.8 | 28.9 | 29.0 | 132.3 | 132.2 | 132.4 | 132.2 | 132.3 | 132.2 | 29.0 | 132.3 | 132.2 |
| 26 | 23.0 | 22.8 | 22.7 | 22.9 | 22.9 | 26.0 | 25.9 | 25.9 | 25.9 | 25.9 | 26.0 | 22.9 | 25.9 | 26.0 |
| 27 | 22.9 | 22.7 | 22.6 | 22.8 | 22.9 | 18.3 | 18.1 | 18 | 18.1 | 18.2 | 18.2 | 23.0 | 18.0 | 18.2 |
| Glc | Glc | Glc | Glc | Glc | Rha | Glc | Glc | Glc | Glc | Glc | Glc | Rha | Glc | |
| 1 | 102.4 | 104.9 | 102.1 | 102.3 | 102.4 | 101.4 | 102.3 | 102.4 | 102.3 | 102.3 | 100.3 | 102.6 | 104.9 | 102.5 |
| 2 | 75.4 | 75.3 | 75.1 | 75.3 | 75.4 | 74.2 | 75.3 | 75.4 | 75.3 | 75.3 | 77.9 | 75.4 | 72.6 | 75.3 |
| 3 | 78.6 | 78.5 | 78.3 | 78.5 | 78.6 | 70.7 | 78.5 | 78.6 | 78.5 | 78.6 | 79.7 | 78.7 | 73.1 | 78.6 |
| 4 | 71.7 | 71.6 | 71.4 | 71.6 | 71.8 | 74.4 | 71.6 | 71.7 | 71.6 | 71.6 | 71.8 | 71.8 | 73.9 | 71.6 |
| 5 | 78.5 | 78.4 | 78.2 | 78.4 | 78.4 | 70.9 | 78.4 | 78.5 | 78.4 | 78.4 | 78.2 | 78.5 | 70.9 | 78.4 |
| 6 | 62.8 | 62.7 | 62.5 | 62.6 | 62.8 | 18.2 | 62.7 | 62.8 | 62.7 | 62.7 | 62.6 | 62.9 | 18.4 | 62.7 |
| Rha | Rha | Rha | Rha | Rha | Rha | Rha | Rha | Rha | Rha | Rha | Rha | |||
| 1 | 101.5 | 102.3 | 101.0 | 101.2 | 101.2 | 101.1 | 104.9 | 101.1 | 101.1 | 101.2 | 101.2 | 101.1 | ||
| 2 | 74.2 | 72.6 | 71.1 | 71.4 | 71.5 | 71.4 | 72.6 | 71.6 | 71.6 | 71.5 | 71.4 | 71.4 | ||
| 3 | 70.7 | 73.1 | 72.6 | 73.1 | 73.8 | 72.8 | 73.1 | 73.1 | 73.7 | 72.9 | 72.9 | 72.8 | ||
| 4 | 74.1 | 73.9 | 70.6 | 71.1 | 71.3 | 70.9 | 74.0 | 71.2 | 71.3 | 71.0 | 71.0 | 70.9 | ||
| 5 | 71.0 | 70.9 | 70.7 | 71.0 | 71.1 | 70.9 | 71.0 | 71.0 | 71.0 | 71.0 | 71.0 | 70.9 | ||
| 6 | 18.3 | 18.4 | 17.9 | 18.2 | 18.2 | 18.1 | 18.4 | 18.2 | 18.2 | 18.2 | 18.2 | 18.1 | ||
| Rha' | ||||||||||||||
| 1 | 102.1 | |||||||||||||
| 2 | 72.6 | |||||||||||||
| 3 | 72.9 | |||||||||||||
| 4 | 74.2 | |||||||||||||
| 5 | 69.5 | |||||||||||||
| 6 | 18.7 | |||||||||||||
| Ac | 21.0 | 20.5 | 20.7 | 20.8 | 21.0 | 20.7 | 20.7 | 20.8 | 20.8 | 20.8 | 20.7 | |||
| 170.4 | 170.2 | 169.9 | 170.0 | 170.6 | 170.2 | 170.0 | 170.1 | 170.1 | 170.2 | 170.2 | ||||
| Ac | 20.6 | 20.8 | 20.9 | 20.8 | 20.8 | |||||||||
| 170.0 | 170.5 | 170.4 | 170.4 | 170.5 | ||||||||||
| PMBz | TMBz | PMBz | TMBz | |||||||||||
| 1 | 123.1 | 125.9 | 123.5 | 125.8 | ||||||||||
| 2 | 132.0 | 107.8 | 132.0 | 107.6 | ||||||||||
| 3 | 114.1 | 153.6 | 114.1 | 153.5 | ||||||||||
| 4 | 163.8 | 143.3 | 163.8 | 143.0 | ||||||||||
| 5 | 114.1 | 153.6 | 114.1 | 153.5 | ||||||||||
| 6 | 132.0 | 107.8 | 132.0 | 107.6 | ||||||||||
| 7 | 165.8 | 165.9 | 165.9 | 166.0 | ||||||||||
| OMe | 55.3 | 56.1 | (x 2) | 55.4 | 55.9 | (x 2) | ||||||||
| OMe | 60.6 | 60.5 |
| Compounds | IC50 (μM) | Compounds | IC50 (μM) |
|---|---|---|---|
| 1 | 10< | 11 | 0.16 ± 0.02 |
| 2 | 10< | 12 | 0.08 ± 0.01 |
| 3 | 1.57 ± 0.10 | 13 | 10< |
| 4 | 10< | 14 | 10< |
| 5 | 10< | 15 | 7.12 ± 0.27 |
| 6 | 10< | 16 | 0.06 ± 0.002 |
| 6a | 10< | 17 | 0.05 ± 0.01 |
| 7 | 7.72 ± 0.42 | 18 | 10< |
| 8 | 5.33 ± 0.33 | 19 | 2.67 ± 0.31 |
| 9 | 5.94 ± 0.10 | etoposide | 0.23 ± 0.02 |
| 10 | 0.09 ± 0.01 | cisplatin | 1.52 ± 0.09 |
| Compounds | IC50 (μM) | Compounds | IC50 (μM) |
|---|---|---|---|
| 10 | 2.41 ± 0.17 | 17 | 0.27 ± 0.04 |
| 11 | 1.84 ± 0.31 | cisplatin | 2.27 ± 0.39 |
| 12 | 0.98 ± 0.09 | doxorubicin | 0.17 ± 0.02 |
| 16 | 0.37 ± 0.09 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iguchi, T.; Kuroda, M.; Naito, R.; Watanabe, T.; Matsuo, Y.; Yokosuka, A.; Mimaki, Y. Structural Characterization of Cholestane Rhamnosides from Ornithogalum saundersiae Bulbs and Their Cytotoxic Activity against Cultured Tumor Cells. Molecules 2017, 22, 1243. https://doi.org/10.3390/molecules22081243
Iguchi T, Kuroda M, Naito R, Watanabe T, Matsuo Y, Yokosuka A, Mimaki Y. Structural Characterization of Cholestane Rhamnosides from Ornithogalum saundersiae Bulbs and Their Cytotoxic Activity against Cultured Tumor Cells. Molecules. 2017; 22(8):1243. https://doi.org/10.3390/molecules22081243
Chicago/Turabian StyleIguchi, Tomoki, Minpei Kuroda, Rei Naito, Tomoyuki Watanabe, Yukiko Matsuo, Akihito Yokosuka, and Yoshihiro Mimaki. 2017. "Structural Characterization of Cholestane Rhamnosides from Ornithogalum saundersiae Bulbs and Their Cytotoxic Activity against Cultured Tumor Cells" Molecules 22, no. 8: 1243. https://doi.org/10.3390/molecules22081243
APA StyleIguchi, T., Kuroda, M., Naito, R., Watanabe, T., Matsuo, Y., Yokosuka, A., & Mimaki, Y. (2017). Structural Characterization of Cholestane Rhamnosides from Ornithogalum saundersiae Bulbs and Their Cytotoxic Activity against Cultured Tumor Cells. Molecules, 22(8), 1243. https://doi.org/10.3390/molecules22081243