Interaction of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods

Abstract
:
1. Introduction
2. Results and Discussion
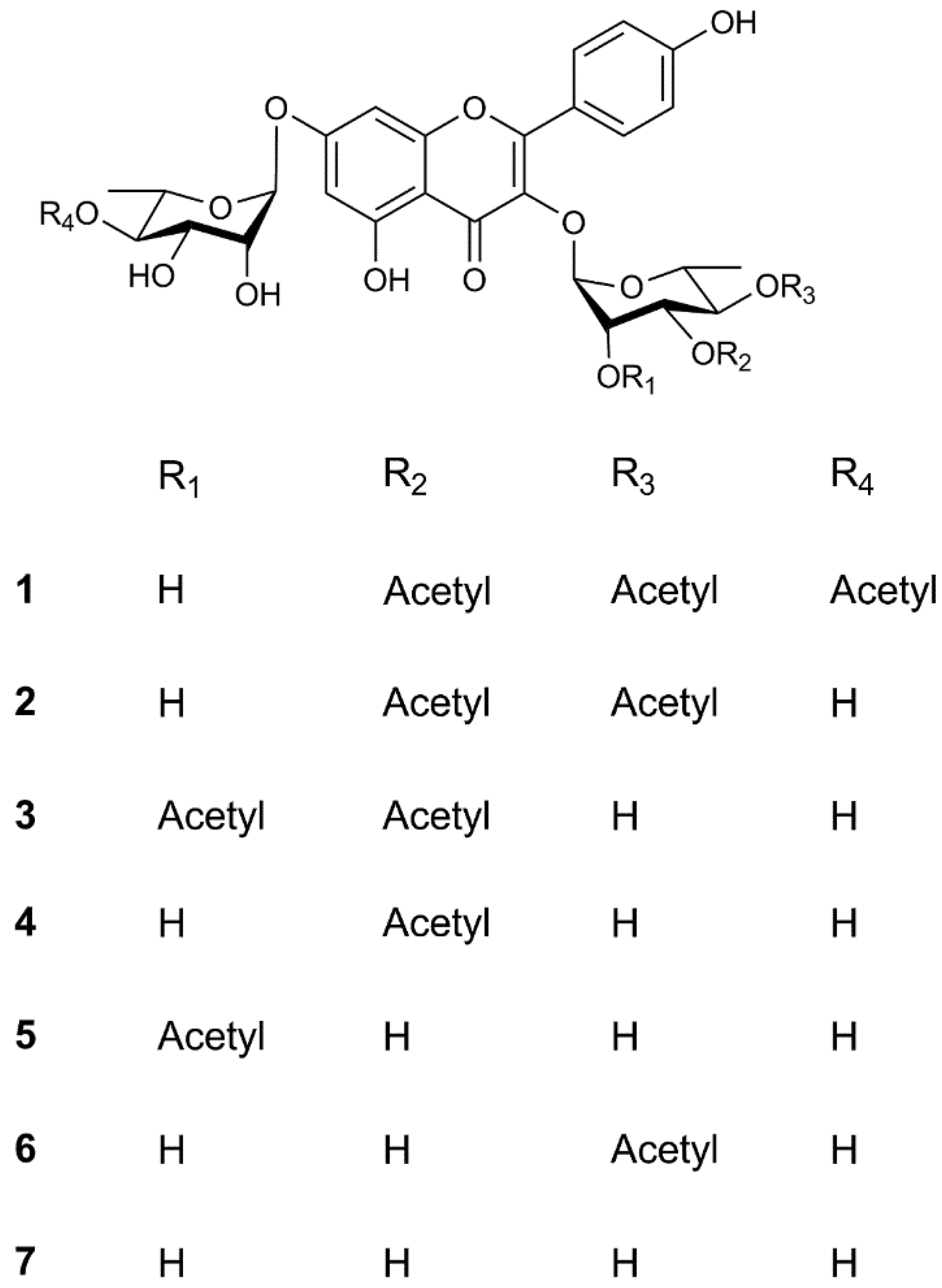
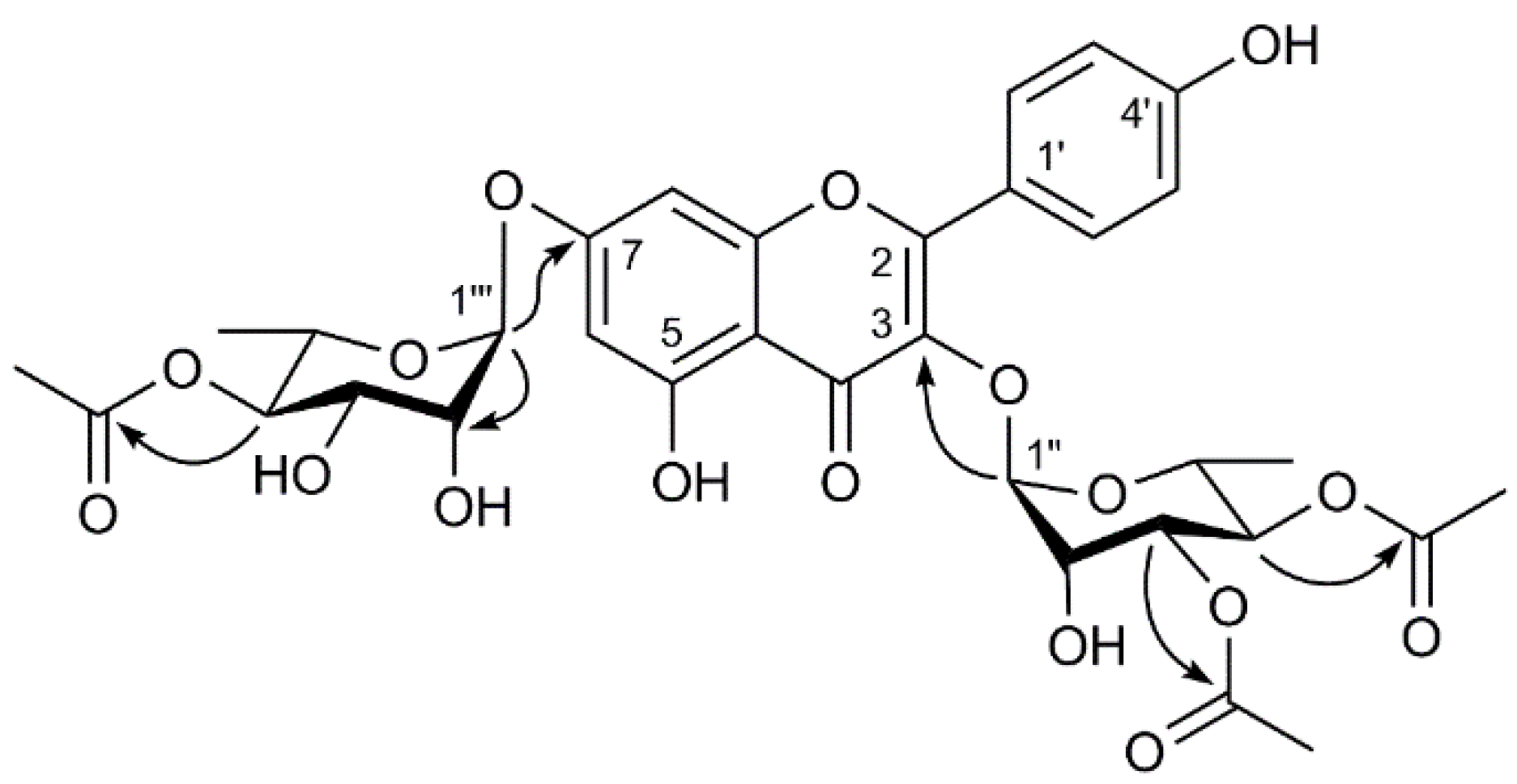
2.1. Structure Elucidation of Compounds 1–7
2.2. Pharmacological Studies
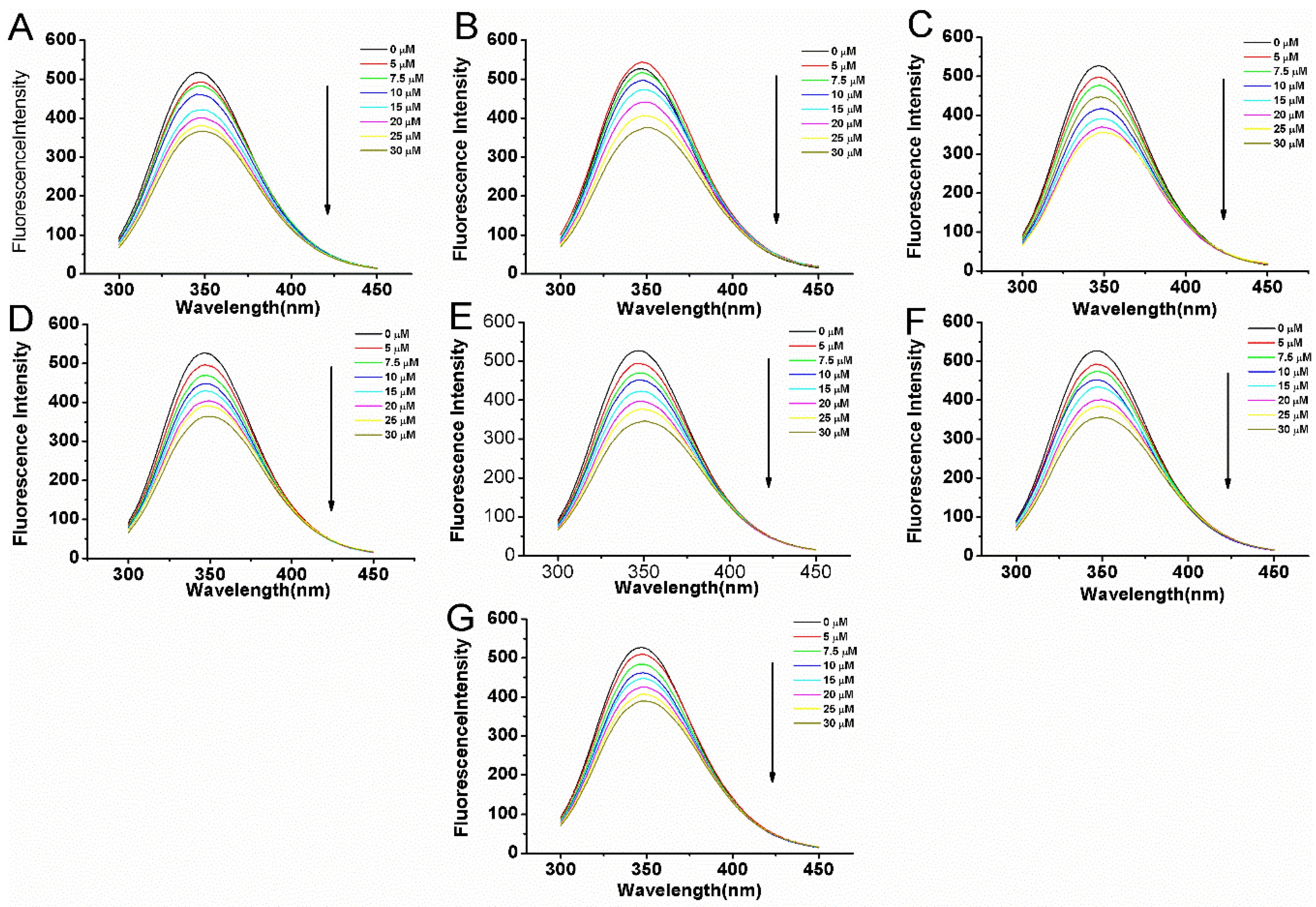
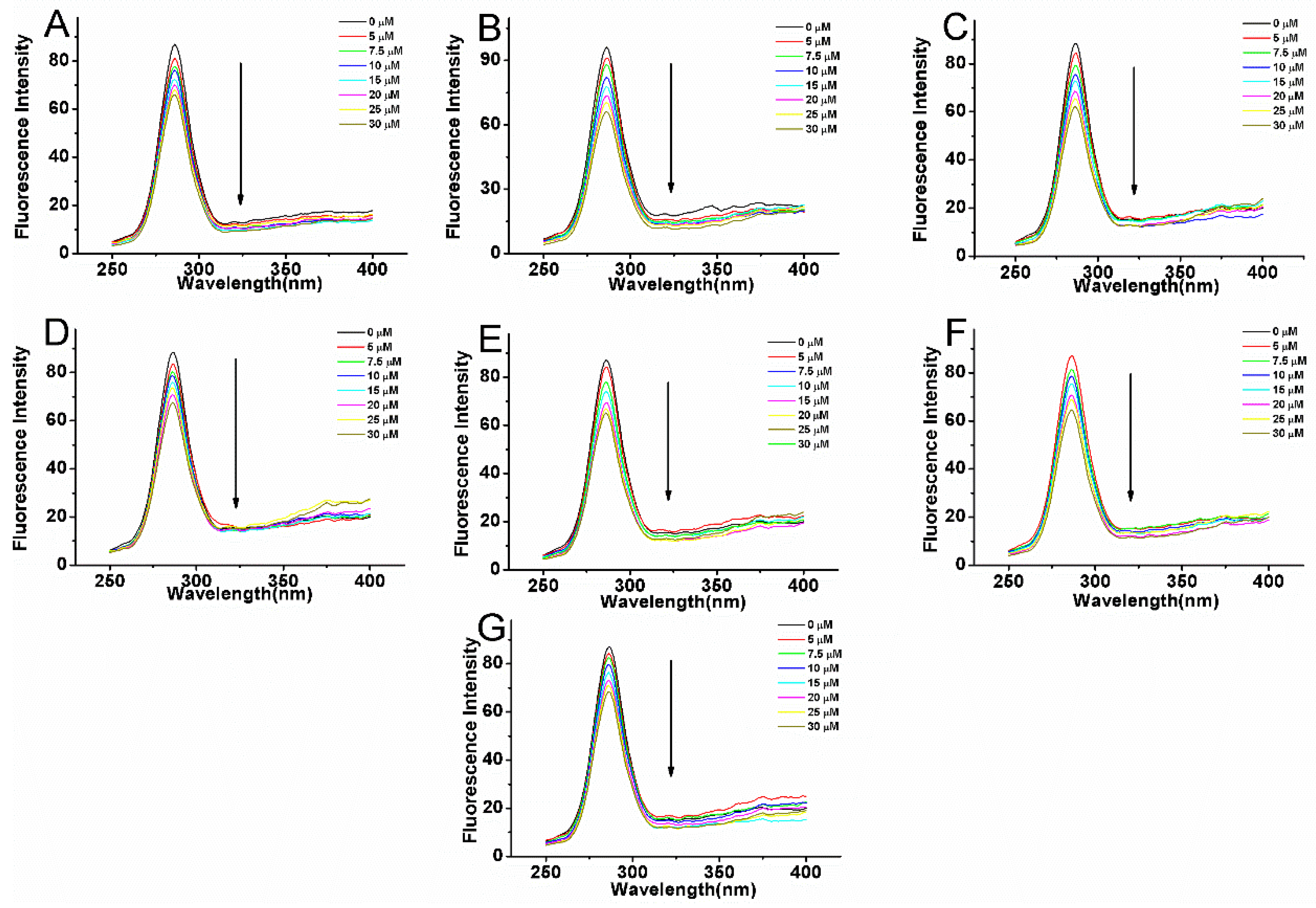
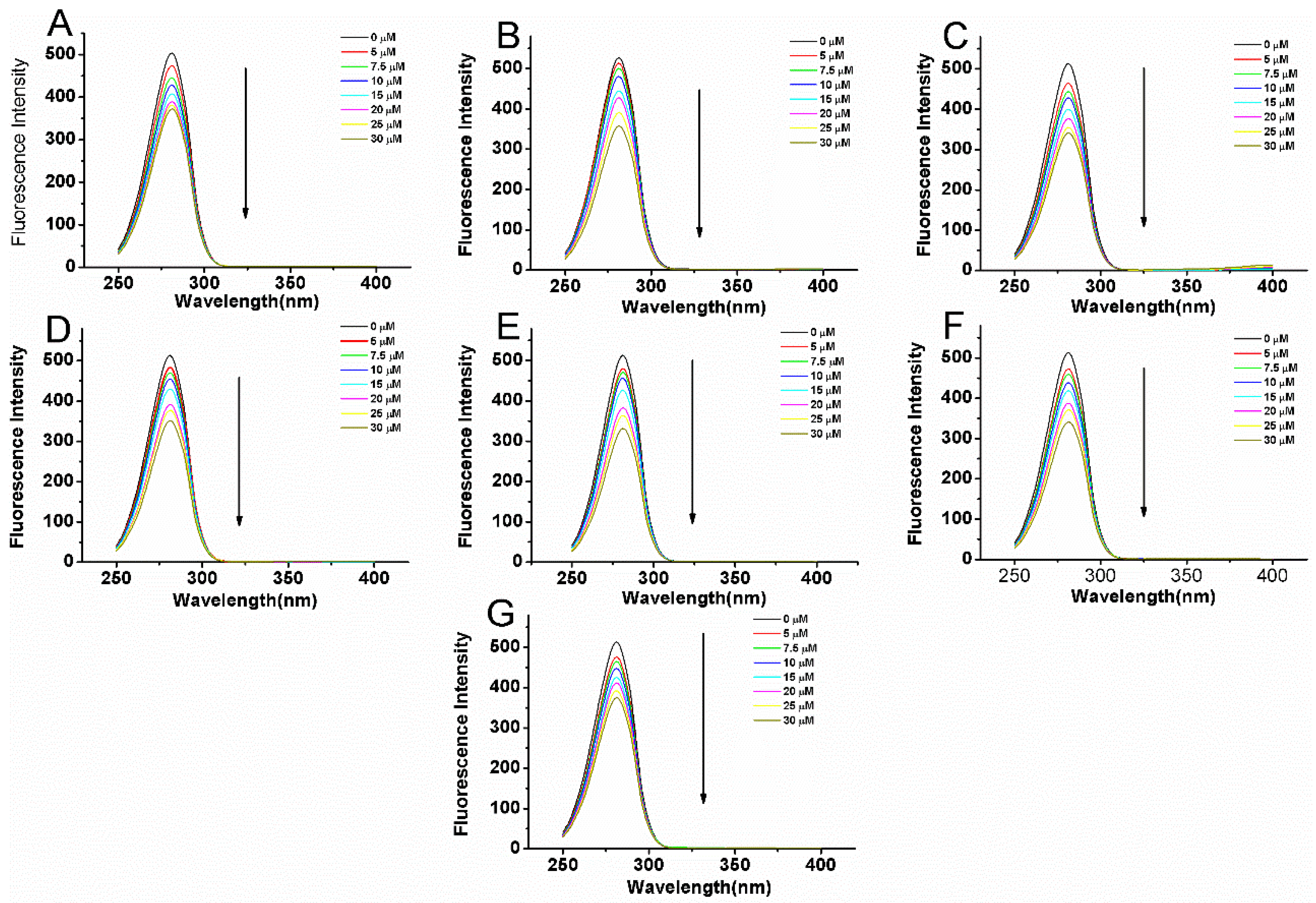
2.3. Fluorescence Quenching of BSA Induced by Compounds
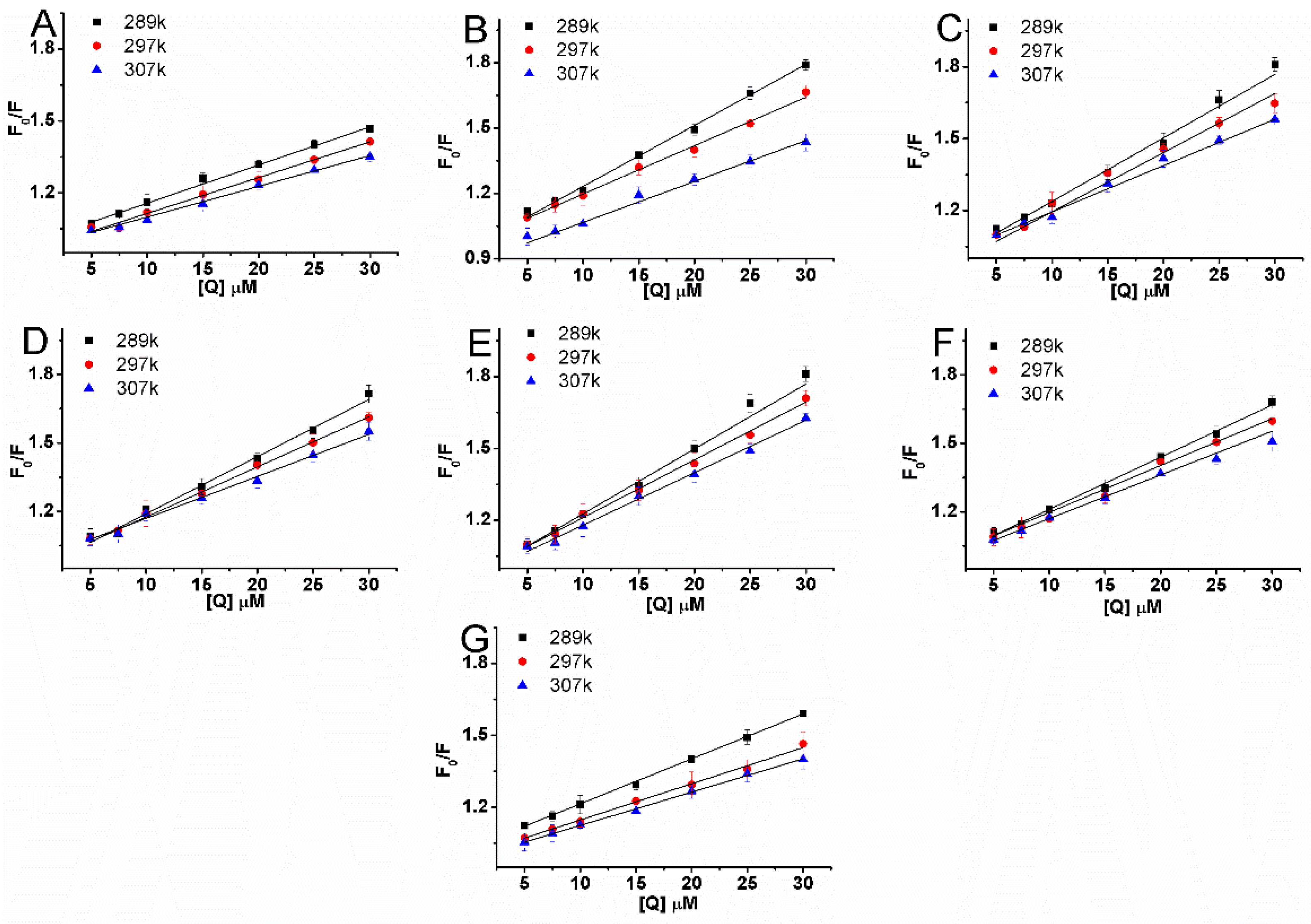
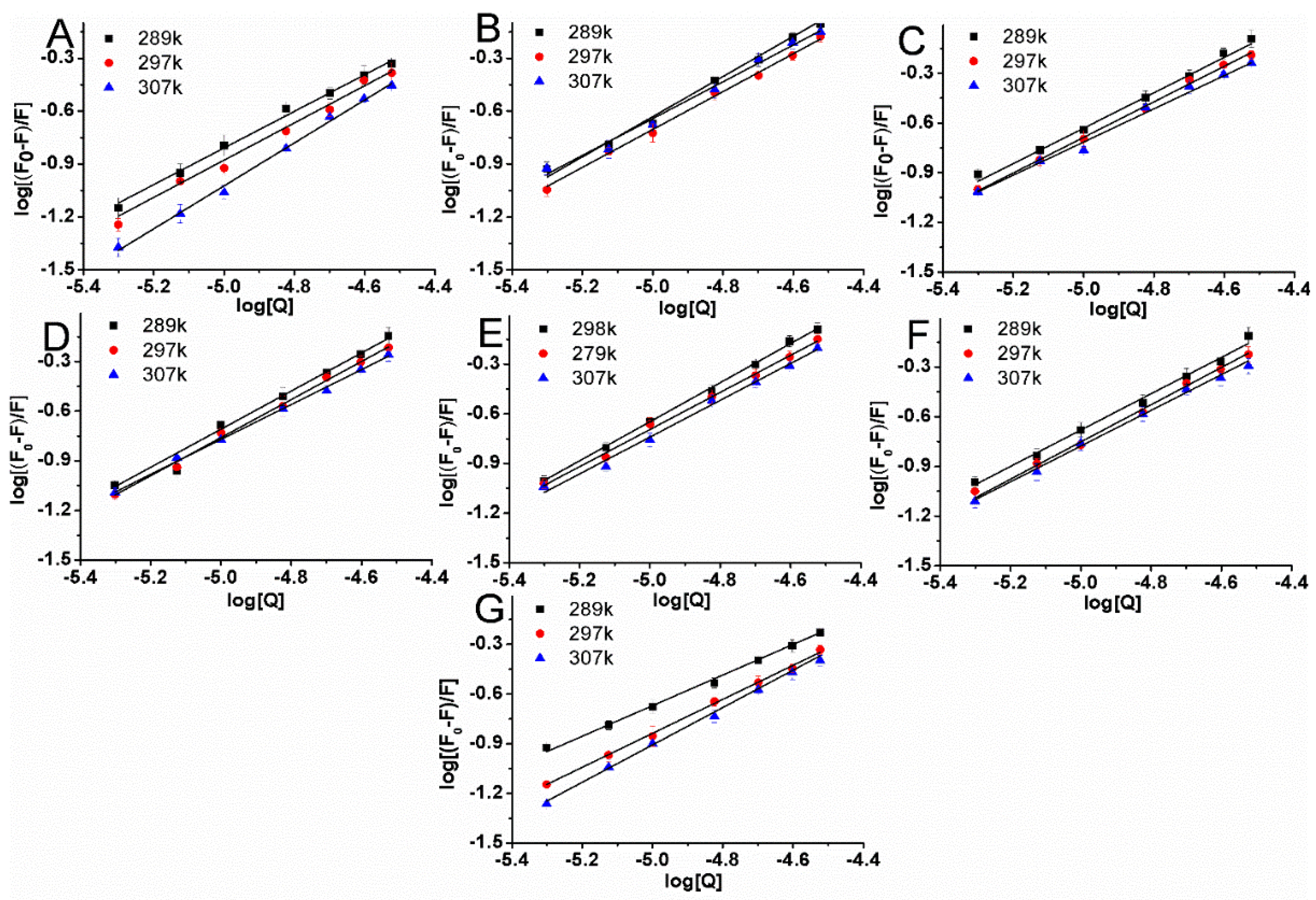
2.4. Types of Interaction Forces between BSA and Compounds
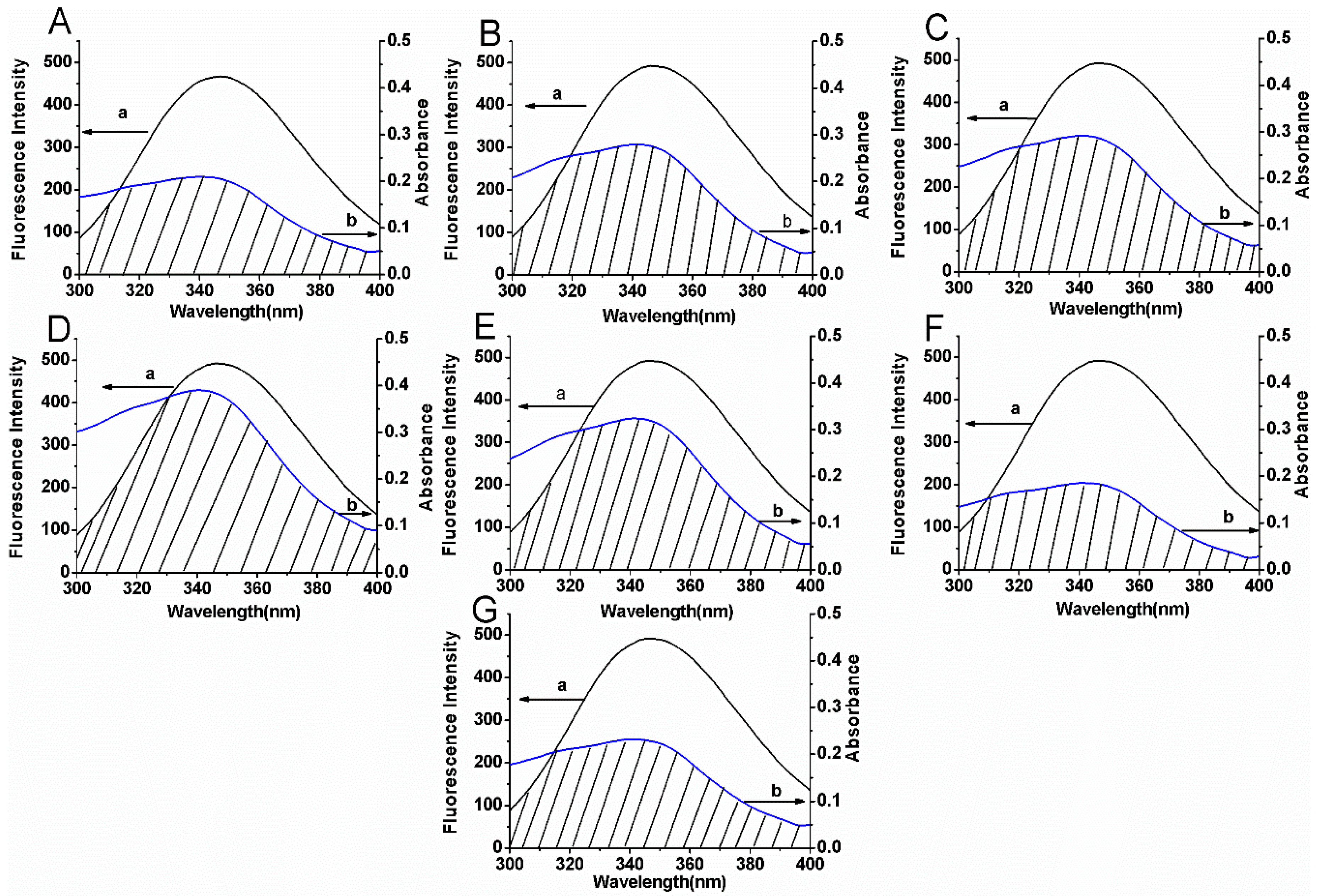
2.5. Synchronous Fluorescence Spectroscopic Studies
2.6. Circular Dichroism Studies
2.7. Energy Transfer from BSA to Compounds
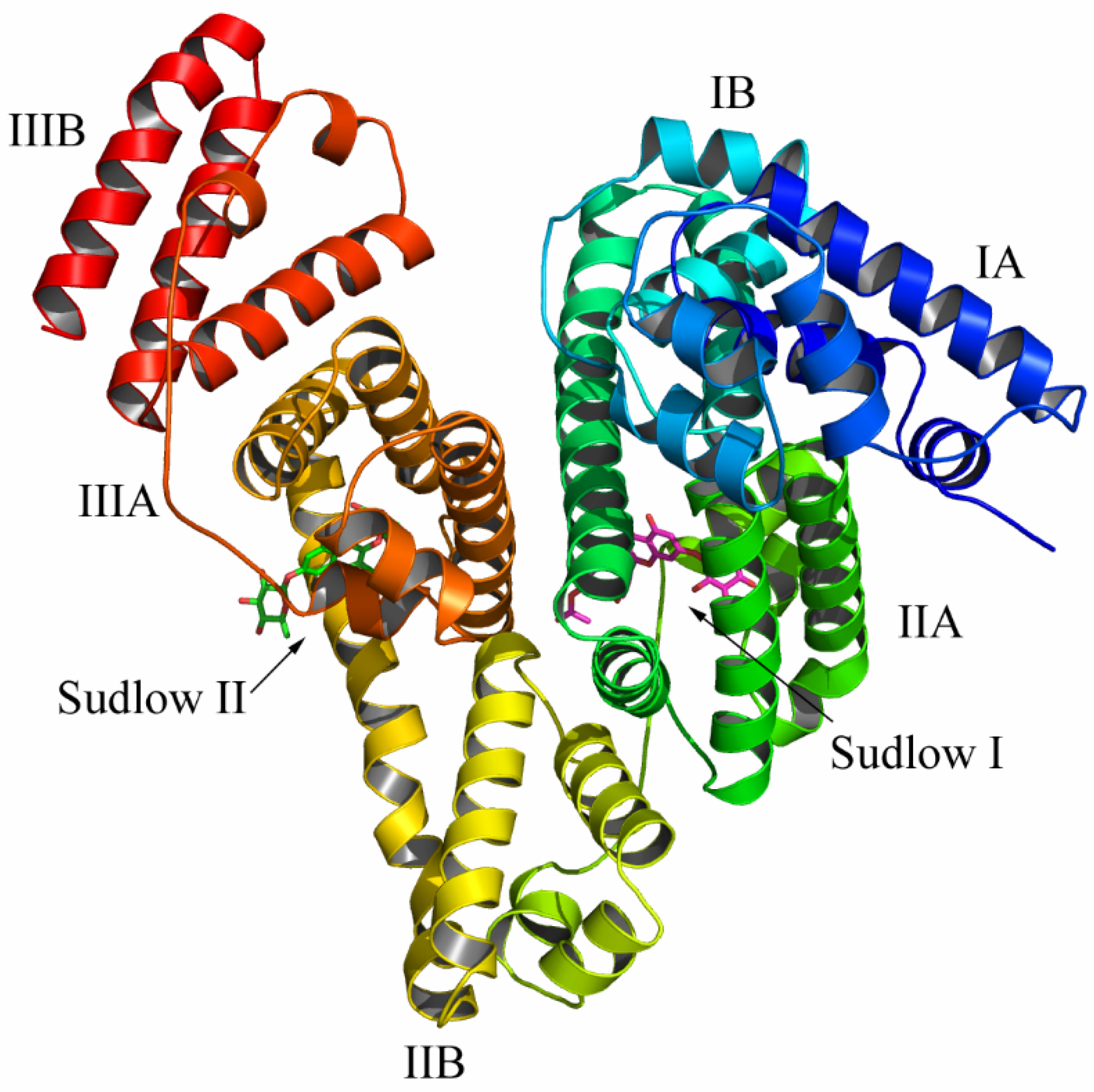
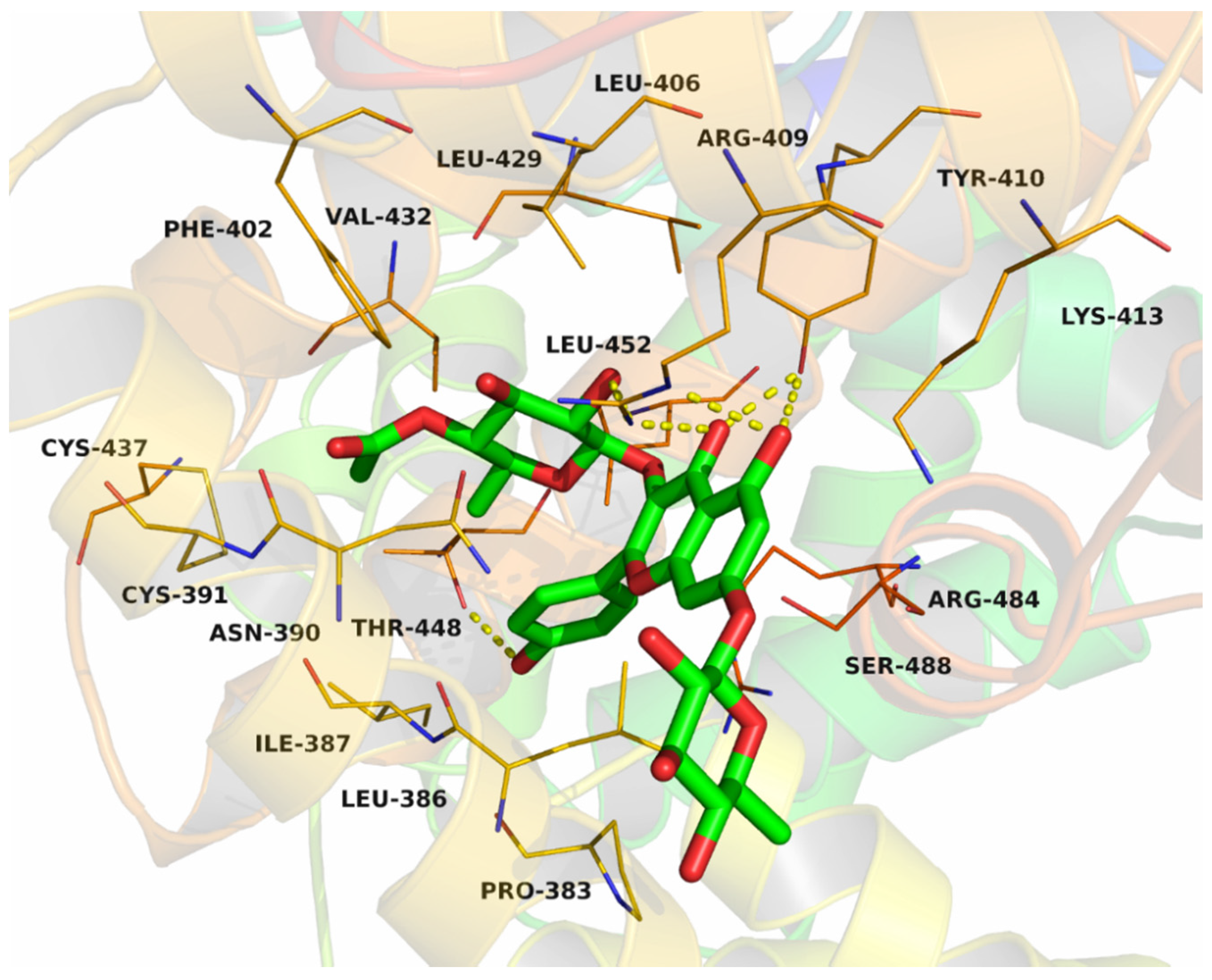
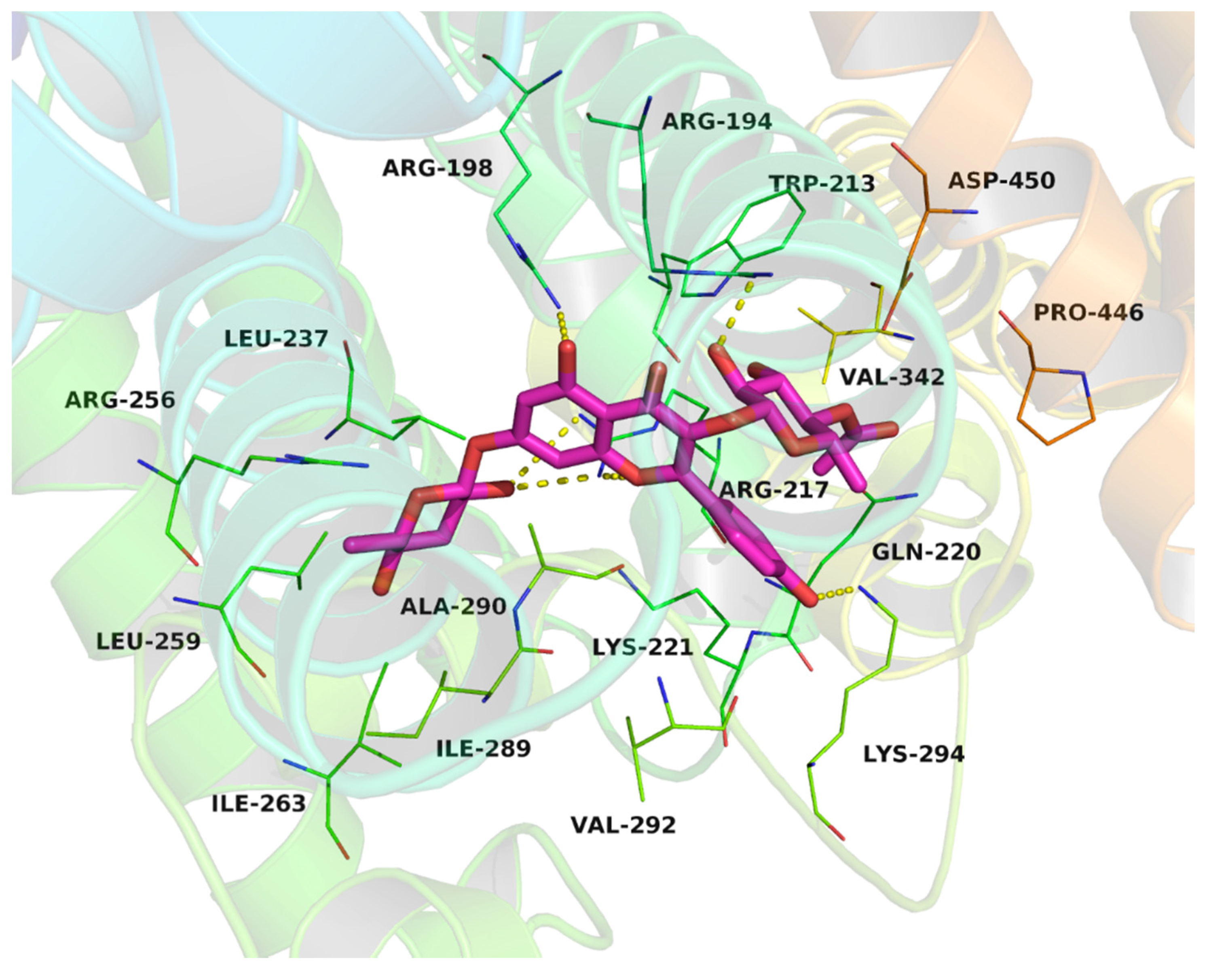
2.8. The determination of Binding Sites of Compound 6 on BSA
3. Materials and Methods
3.1. Chemicals and Instrumentation
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Sugar Identification
3.5. MTT Cytotoxicity Assay
3.6. BSA Binding Experiment
3.7. Molecule Modeling
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Q.; Feng, Y.; Kennedy, D.; Kennedy, D. Multidrug-resistant cancer cells and cancer stem cells hijack cellular systems to circumvent systemic therapies, can natural products reverse this? Cell. Mol. Life Sci. 2017, 74, 777–801. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.Y.; Gu, Y.H.; Cao, C.J.; Wang, J.; Han, D.D.; Tang, Y.C.; Chen, H.S.; Xu, A. Reversal effect and mechanism of Ginkgo biloba exocarp extracts in multidrug resistance of mice S180 tumor cells. Exp. Ther. Med. 2016, 12, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Kim, Y.; Choo, H.; Chong, Y. Quercetin-glutamic acid conjugate with a non-hydrolysable linker; a novel scaffold for multidrug resistance reversal agents through inhibition of P-glycoprotein. Bioorg. Med. Chem. 2017, 25, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Poureshghi, F.; Ghandforoushan, P.; Safarnejad, A.; Soltani, S. Interaction of an antiepileptic drug, lamotrigine with human serum albumin (HSA): Application of spectroscopic techniques and molecular modeling methods. J. Photochem. Photobiol. 2017, 166, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.Y.; Zhou, K.L.; Pan, D.Q.; Shen, J.L.; Shi, J.L. Spectroscopic and molecular docking approaches for investigating conformation and binding characteristics of clonazepam with bovine serum albumin (BSA). J. Photochem. Photobiol. 2017, 167, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.Q.; Jiang, M.; Liu, T.T.; Wang, Q.; Shi, J.H. Combined spectroscopies and molecular docking approach to characterizing the binding interaction of enalapril with bovine serum albumin. Luminescence 2017, 32, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hanuš, L.O.; Řezanka, T.; Dembitsky, V.M. A trinorsesterterpene glycoside from the North American fern Woodwardia virginica (L.) Smith. Phytochemistry 2003, 63, 869–875. [Google Scholar] [CrossRef]
- Řezanka, T.; Dembitsky, V.M.; Hanuš, L.O. Two cyclohexenone glycosides from the North American fern Woodwardia virginica (L.) Smith. Phytochemistry 2003, 63, 931–937. [Google Scholar] [CrossRef]
- Wang, S.Q.; Li, X.; Wang, X.N.; Wei, N.N.; Lou, H.X. Coumarins from Cicuta virosa and their modulating effects on multidrug-resistant (MDR) tumors. Phytochem. Lett. 2011, 4, 97–100. [Google Scholar] [CrossRef]
- Wang, S.Q.; Wang, X.N.; Li, Y.Y.; Di, X.X.; Lou, H.X. Identification of purine-derived compounds, ustilagomaydisin A-C, from the plant pathogen Ustilago maydis and their modulating effects on multidrug-resistant (MDR) tumors. Phytochem. Lett. 2014, 10, 193–197. [Google Scholar] [CrossRef]
- Diaz, J.G.; Herz, W. Acylated flavonol tetraglycosides from Delphinium gracile. Phytochemistry 2010, 71, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Ren, D.M.; Xiang, F.; Wang, X.N.; Zhu, C.J.; Yuan, H.Q.; Sun, L.M.; Lv, B.B.; Sun, X.J.; Lou, H.X. Dracotanosides A-D, spermidine glycosides from Dracocephalum tanguticum: Structure and amide rotational barrier. J. Nat. Prod. 2009, 72, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Zhu, X.F.; Wang, X.N.; Shen, T.; Xiang, F.; Lou, H.X. Flavonoids from Malus hupehensis and their cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells. Phytochemistry 2013, 87, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Kondratyuk, T.P.; Marler, L.E.; Qiu, X.; Choi, Y.; Cao, H.; Yu, R.; Sturdy, M.; Pegan, S.; Liu, Y.; et al. Isolation and evaluation of kaempferol glycosides from the fern Neocheiropteris palmatopedata. Phytochemistry 2010, 71, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Miyashiro, H.; Nakamura, N.; Hattori, M. Two new triterpenes from the Rhizome of Dryopteris crassirhizoma, and inhibitory activities of its constituents on human immunodeficiency virus-1 protease. Chem. Pharm. Bull. 2008, 56, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Spanou, C.; Bourou, G.; Dervishi, A.; Aligiannis, N.; Angelis, A.; Komiotis, D.; Skaltsounis, A.L.; Kouretas, D. Antioxidant and chemopreventive properties of polyphenols compounds derived from Greek legume plant extracts. J. Agric. Food Chem. 2008, 56, 6967–6976. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Iinuma, M.; Tanaka, T.; Yamamoto, H. Products, Sutchuenoside A: A new kaempferol glycoside from the aerial parts of Epimedium sutchuenense. J. Nat. Prod. 1991, 54, 1427–1429. [Google Scholar] [CrossRef]
- Li, M.; Huang, S.; Ye, C.; Xie, Y. Synthesis, structure, protein binding of Cu(II) complexes with a tridentate NNO Schiff-base ligand. Spectrochim. Acta A 2015, 150, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Klotz, I.M. Physiochemical aspects of drug-protein interactions: a general perspective. Ann. N. Y. Acad. Sci. 1973, 226, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Gentili, P.L.; Ortica, F.; Favaro, G.; Chimica, D.; Uni, V. Static and dynamic interaction of a naturally occurring photochromic molecule with bovine serum albumin studied by UV-Visible absorption and Fluorescence Spectroscopy. J. Phys. Chem. B 2008, 112, 16793–16801. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Liu, Y.; Xie, M.X.; Li, S.; Jiang, M.; Wang, Y.D. Interactions of human serum albumin with chlorogenic acid and ferulic acid. Acta (BBA) Gen. Subj. 2004, 1674, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhu, M.; Xu, C.; Ji, B. Characterization of the baicalein-bovine serum albumin complex without or with Cu2+or Fe3+ by spectroscopic approaches. Eur. J. Med. Chem. 2011, 46, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Safarnejad, A.; Shaghaghi, M.; Dehghan, G.; Soltani, S. Binding of carvedilol to serum albumins investigated by multi-spectroscopic and molecular modeling methods. J. Lumin. 2016, 176, 149–158. [Google Scholar] [CrossRef]
- Śliwińska-Hill, U. Interaction of imatinib mesylate with human serum transferrin: The comparative spectroscopic studies. Spectrochim. Acta A 2017, 173, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Piszczek, G.; Kang, J.S. On the possibility of long-wavelength long lifeti high-quantum-yield luminopores. Anal. Biochem. 2001, 288, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Il′ichev, Y.V.; Perry, J.L.; Simon, J.D. Interaction of ochratoxin A with human serum albumin. Preferential binding of the dianion and pH effects. J. Phy. Chem. B 2001, 106, 452–459. [Google Scholar] [CrossRef]
- Foresman, J.B.A. Frisch, Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Sekula, B.; Zielinski, K.; Bujacz, A. Crystallographic studies of the complexes of bovine and equine serum albumin with 3,5-diiodosalicylic acid. Int. J. Biol. Macromol. 2013, 60, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Pan, D.Q.; Wang, X.X.; Liu, T.T.; Jiang, M.; Wang, Q. Characterizing the binding interaction between antimalarial artemether (AMT) and bovine serum albumin (BSA): Spectroscopic and molecular docking methods. J. Photochem. Photobiol. 2016, 162, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1−7 are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (J in Hz) | δC | HMBC (H → C) |
|---|---|---|---|
| 2 | 158.5 | ||
| 3 | 134.0 | ||
| 4 | 178.1 | ||
| 5 | 161.6 | ||
| 6 | 6.52 d (1.8) | 99.2 | C-5, C-7, C-8 |
| 7 | 162.2 | ||
| 8 | 6.79 d (1.8) | 94.2 | C-7, C-10, C-6 |
| 9 | 156.7 | ||
| 10 | 106.2 | ||
| 1′ | 120.8 | ||
| 2′ | 7.82 d (8.4) | 130.6 | C-1′, C-3′/5′, C-4′ |
| 3′ | 7.00 d (8.4) | 115.3 | C-1′, C-2′/6′, C-4′ |
| 4′ | 160.6 | ||
| 5′ | 7.00 d (8.4) | 115.3 | C-1′, C-2′/6′, C-4′ |
| 6′ | 7.82 d (8.4) | 130.6 | C-1′, C-3′/5′, C-4′ |
| 1′′ | 5.64 d (1.2) | 100.5 | C-3, C-2′′ |
| 2′′ | 4.36 m | 67.7 | C-1′′, C-3′′ |
| 3′′ | 5.18 dd (10.2 3.0) | 71.3 | C-4′′, 3′′-COCH3 |
| 4′′ | 5.05 t (10.2) | 70.3 | C-5′′, 4′′-COCH3 |
| 5′′ | 3.78 m | 68.3 | C-4′′, C-6′′ |
| 6′′ | 0.84 d(6.0) | 16.1 | C-4′′, C-5′′ |
| 1′′′ | 5.58 m | 98.5 | C-7, C-2′′ |
| 2′′′ | 4.24 m | 73.3 | C-1′′′, C-3′′′, |
| 3′′′ | 4.06 dd (9.6 3.0) | 70.4 | C-2′′′, C-4′′′ |
| 4′′′ | 4.78 t (9.6) | 73.3 | C-3′′′, 4′′′-COCH3 |
| 5′′′ | 3.33 m | 69.9 | C-4′′′, C-6′′′ |
| 6′′′ | 1.17 d (6.0) | 16.7 | C-5′′′ |
| 3′′-COCH3 | 170.7 | ||
| 3′′-COCH3 | 2.10 s | 19.4 | 3′′-COCH3 |
| 4′′-COCH3 | 170.3 | ||
| 4′′-COCH3 | 2.00 s | 19.2 | 4′′-COCH3 |
| 4′′′-COCH3 | 170.9 | ||
| 4′′′-COCH3 | 1.96 s | 19.5 | 4′′′-COCH3 |
| Compounds | IC50 (K562) | IC50 (K562/A02) | RF (K562/A02) |
|---|---|---|---|
| DOX | 0.66 ± 0.12 | 19.2 ± 0.31 | |
| DOX + Verapamil | 0.58 ± 0.23 | 2.12 ± 0.19 | 9.06 |
| DOX + 1 | 0.67 ± 0.11 | 5.18 ± 0.17 | 3.71 |
| DOX + 2 | 0.62 ± 0.08 | 4.14 ± 0.33 | 4.64 |
| DOX + 3 | 0.69 ± 0.10 | 3.74 ± 0.12 | 5.13 |
| DOX + 4 | 0.70 ± 0.24 | 3.04 ± 0.33 | 4.75 |
| DOX + 5 | 0.65 ± 0.16 | 5.14 ± 0.12 | 3.74 |
| DOX + 6 | 0.68 ± 0.12 | 2.31 ± 0.10 | 8.31 |
| DOX + 7 | 0.60 ± 0.22 | 3.93 ± 0.26 | 5.94 |
| NO | T (K) | Ksv (×104 L·mol−1) | R2 | Kq (×1012 L·mol−1·s−1) | Log Kb | Kb (×105 L·mol−1) | n | R2 | ΔG (KJ·mol−1) | ΔH (KJ·mol−1) | ΔS (J·mol−1·K−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 289 | 1.6 | 0.994 | 1.6 | 4.472 | 0.3 | 1.06 | 0.994 | |||
| 297 | 1.49 | 0.991 | 1.49 | 4.616 | 0.41 | 1.1 | 0.992 | −26.25 | 67.1 | 316.68 | |
| 307 | 1.3 | 0.995 | 1.3 | 5.082 | 1.21 | 1.22 | 0.996 | ||||
| 2 | 289 | 2.76 | 0.996 | 2.76 | 5.332 | 2.15 | 1.1 | 0.986 | |||
| 297 | 2.24 | 0.995 | 2.24 | 4.757 | 0.57 | 1.09 | 0.997 | −27.05 | −84.47 | −191.14 | |
| 307 | 1.79 | 0.993 | 1.79 | 4.589 | 0.39 | 1.06 | 0.991 | ||||
| 3 | 289 | 2.77 | 0.993 | 2.77 | 4.701 | 0.5 | 1.06 | 0.995 | |||
| 297 | 2.26 | 0.99 | 2.26 | 4.675 | 0.47 | 1.07 | 0.997 | −26.58 | −29.55 | −11.55 | |
| 307 | 1.99 | 0.992 | 1.99 | 4.43 | 0.27 | 1.03 | 0.991 | ||||
| 4 | 289 | 2.5 | 0.993 | 2.5 | 5.265 | 1.84 | 1.2 | 0.985 | |||
| 297 | 2.14 | 0.998 | 2.14 | 5.027 | 1.06 | 1.16 | 0.996 | −28.58 | −89.64 | −208.18 | |
| 307 | 1.87 | 0.992 | 1.87 | 4.453 | 0.28 | 1.04 | 0.998 | ||||
| 5 | 289 | 2.91 | 0.995 | 2.91 | 5.277 | 1.89 | 1.19 | 0.998 | |||
| 297 | 2.4 | 0.996 | 2.4 | 4.984 | 0.96 | 1.14 | 0/995 | −28.34 | −54.79 | −88.71 | |
| 307 | 2.14 | 0.996 | 2.14 | 4.79 | 0.62 | 1.11 | 0.993 | ||||
| 6 | 289 | 2.29 | 0.996 | 2.29 | 4.894 | 0.78 | 1.08 | 0.997 | |||
| 297 | 2.11 | 0.993 | 2.11 | 4.662 | 0.46 | 1.08 | 0.996 | −26.69 | −38 | −38.1 | |
| 307 | 1.75 | 0.995 | 1.75 | 4.558 | 0.36 | 1.07 | 0.996 | ||||
| 7 | 289 | 2.74 | 0.998 | 2.74 | 3.819 | 0.07 | 0.9 | 0.997 | |||
| 297 | 1.53 | 0.996 | 1.53 | 4.327 | 0.21 | 1.03 | 0.998 | −24.6 | 90.73 | 387.43 | |
| 307 | 1.4 | 0.998 | 1.4 | 4.624 | 0.42 | 1.11 | 0.998 |
| Compounds | J (cm3 L M−1) | R0 (nm) | r (nm) | E (%) |
|---|---|---|---|---|
| 1 | 7.70 × 10−15 | 2.35 | 2.72 | 29.30 |
| 2 | 9.90 × 10−15 | 2.45 | 2.62 | 39.97 |
| 3 | 1.05 × 10−14 | 2.47 | 2.62 | 41.25 |
| 4 | 1.44 × 10−14 | 2.60 | 2.79 | 39.69 |
| 5 | 1.16 × 10−14 | 2.51 | 2.69 | 39.98 |
| 6 | 6.47 × 10−15 | 2.28 | 2.42 | 41.23 |
| 7 | 8.50 × 10−15 | 2.39 | 2.64 | 35.20 |
| System | LogKb | Kb (×105 L mol−1) | R2 |
|---|---|---|---|
| BSA + compound 6 | 4.662 | 0.459 | 0.993 |
| BSA + compound 6 + ibuprofen | 4.324 | 0.211 | 0.989 |
| BSA + compound 6 + warfarin | 3.227 | 0.017 | 0.993 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, R.; Pan, H.; Shen, T.; Li, P.; Chen, Y.; Li, Z.; Di, X.; Wang, S. Interaction of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods. Molecules 2017, 22, 1317. https://doi.org/10.3390/molecules22081317
Ma R, Pan H, Shen T, Li P, Chen Y, Li Z, Di X, Wang S. Interaction of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods. Molecules. 2017; 22(8):1317. https://doi.org/10.3390/molecules22081317
Chicago/Turabian StyleMa, Rui, Hong Pan, Tao Shen, Peng Li, Yanan Chen, Zhenyu Li, Xiaxia Di, and Shuqi Wang. 2017. "Interaction of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods" Molecules 22, no. 8: 1317. https://doi.org/10.3390/molecules22081317
APA StyleMa, R., Pan, H., Shen, T., Li, P., Chen, Y., Li, Z., Di, X., & Wang, S. (2017). Interaction of Flavonoids from Woodwardia unigemmata with Bovine Serum Albumin (BSA): Application of Spectroscopic Techniques and Molecular Modeling Methods. Molecules, 22(8), 1317. https://doi.org/10.3390/molecules22081317