Solution NMR Studies of Mycobacterium tuberculosis Proteins for Antibiotic Target Discovery

Abstract
:1. Introduction
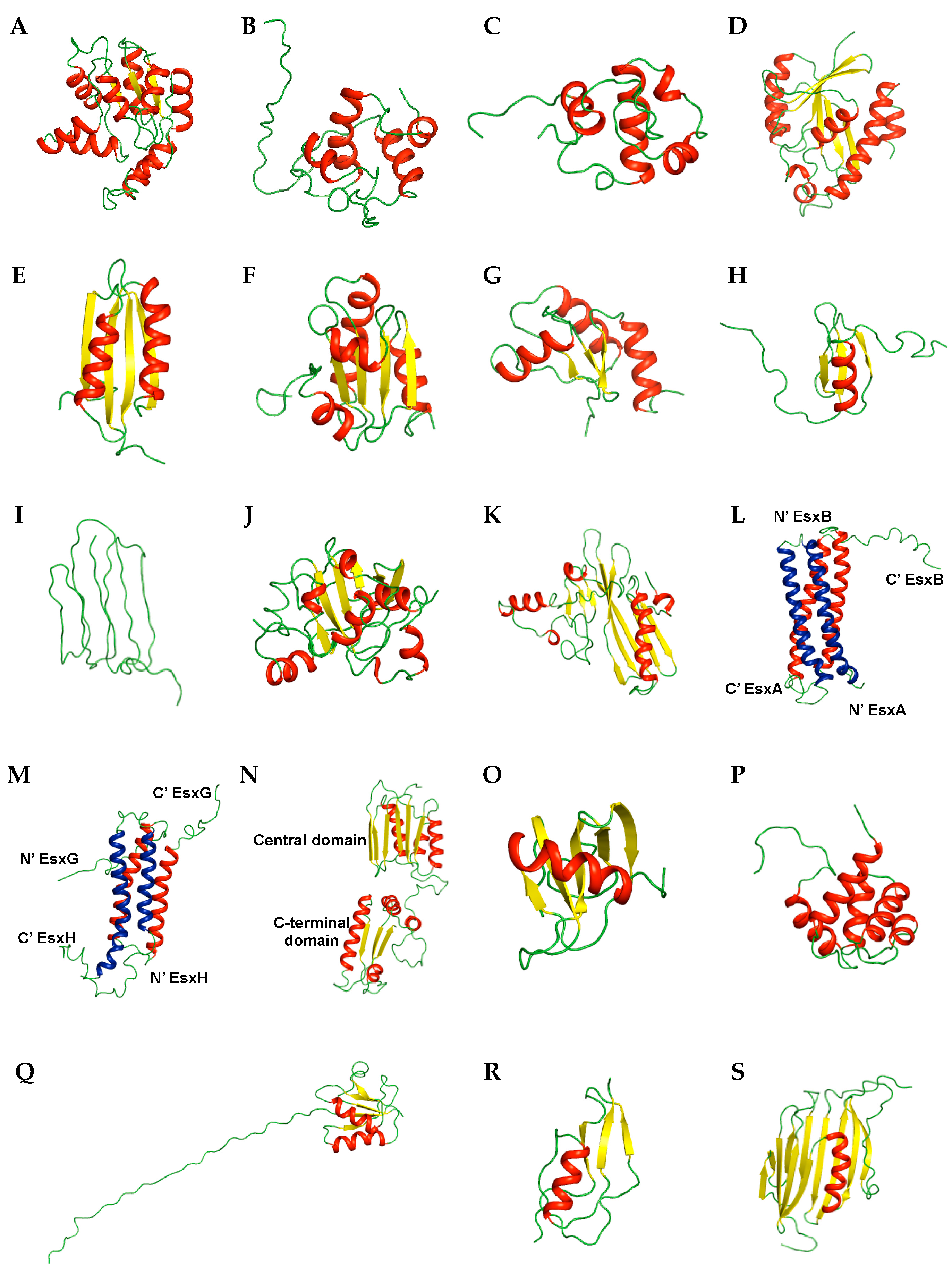
2. NMR Structures
2.1. Transport-Related Proteins
2.2. Transcription-Related Proteins
2.3. Nucleotide-Binding Proteins
2.4. Ser/Thr Protein Kinase-Related Proteins
2.5. Enzymes and Related Proteins
2.6. Siderophore-Related Proteins
2.7. Secreted Proteins
2.8. Membrane Proteins
2.9. Uncharacterized Proteins
2.10. Other Proteins
3. NMR-Based Molecular Interaction
3.1. Protein-Observed NMR for Interaction Mode
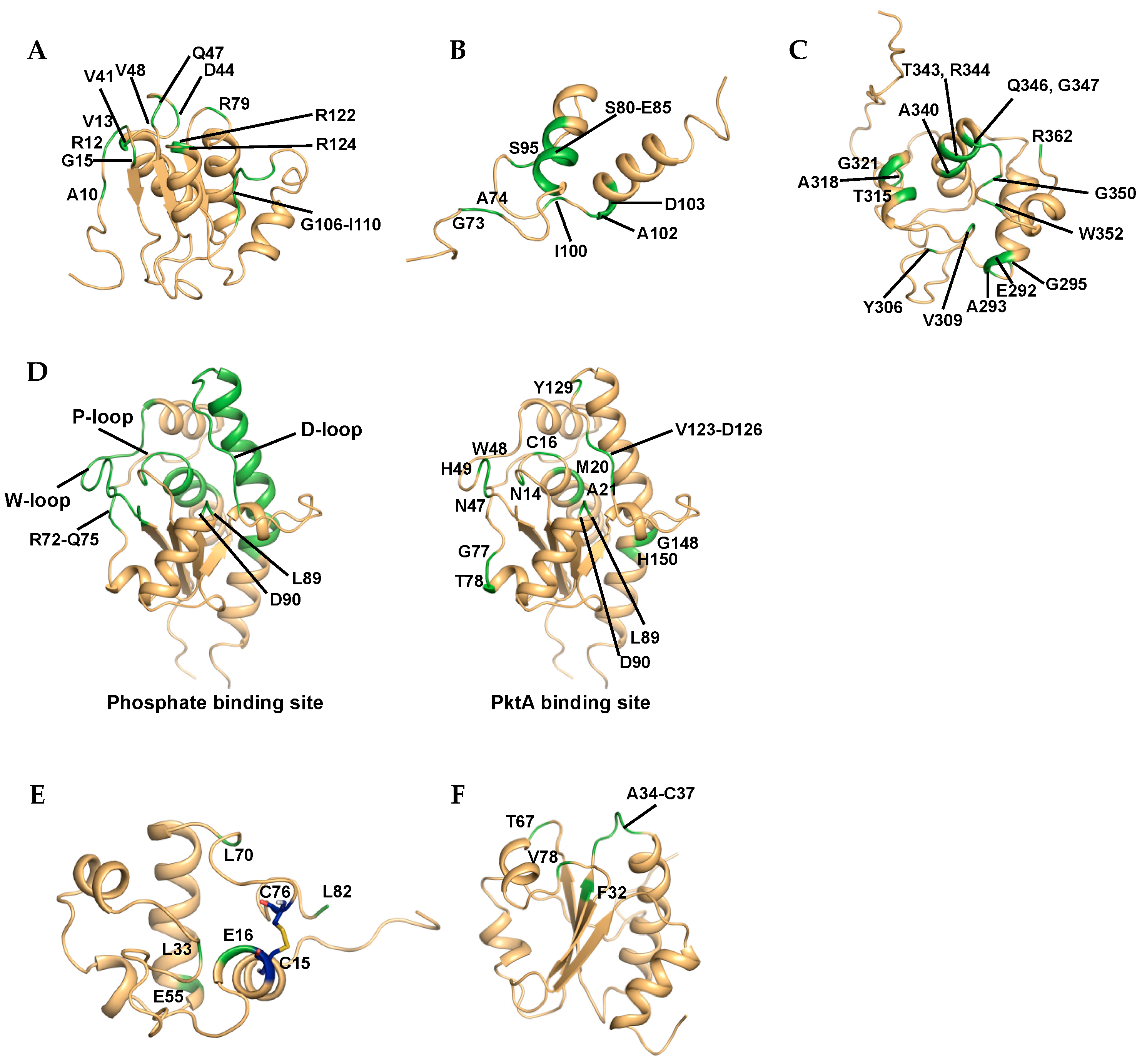
3.1.1. Rv2050, RbpA
3.1.2. Rv1739c
3.1.3. Rv3597c, Lsr2
3.1.4. Rv1009, RpfB
3.1.5. Rv2234, MptpA
3.1.6. Rv1884c, RpfC
3.1.7. Rv3914, TrxC
3.1.8. Rv3682, PonA2
3.2. Ligand-Observed NMR for Drug Discovery
3.2.1. Rv3809c, UDP-Galactopyranose Mutase (UGM)
3.2.2. Rv2276, CYP121
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fu, L.M.; Fu-Liu, C.S. Is Mycobacterium tuberculosis a closer relative to Gram-positive or Gram-negative bacterial pathogens? Tuberculosis 2002, 82, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz, I.; Donoghue, H.D.; Minnikin, D.E.; May, H.; Lee, O.Y.; Feldman, M.; Galili, E.; Spigelman, M.; Rothschild, B.M.; Bar-Gal, G.K. Tuberculosis origin: The Neolithic scenario. Tuberculosis 2015, 95 (Suppl. 1), S122–S126. [Google Scholar] [CrossRef] [PubMed]
- Keri, R.S.; Chand, K.; Ramakrishnappa, T.; Nagaraja, B.M. Recent progress on pyrazole scaffold-based antimycobacterial agents. Arch. Pharm. 2015, 348, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Comolet, T. Multidrug-resistant tuberculosis: Challenges of a global emergence. Bull. Soc. Pathol. Exot. 2015, 108, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Pechalrieu, D.; Lopez, M. Compounds for use in the treatment of mycobacterial infections: A patent evaluation (WO2014049107A1). Exp. Opin. Ther. Pat. 2015, 25, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; de Cesare, M.; et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 10063. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ni, Z.; Shi, Y.; Sun, X.; Wang, H.; Wei, C.; Wang, G.; Li, F. Screening essential genes of Mycobacterium tuberculosis with the pathway enrichment method. Mol. Biol. Rep. 2014, 41, 7639–7644. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.C.; Liu, G.; Zhang, Y.M.; Rock, C.O.; Zheng, J. The solution structure of acyl carrier protein from Mycobacterium tuberculosis. J. Biol. Chem. 2002, 277, 15874–15880. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.O.; Cronan, J.E. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim. Biophys. Acta 1996, 1302, 1–16. [Google Scholar] [CrossRef]
- Brennan, P.J.; Nikaido, H. The envelope of mycobacteria. Annu. Rev. Biochem. 1995, 64, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.L.; Presnell, S.R.; Cohen, F.E. Four helix bundle diversity in globular proteins. J. Mol. Biol. 1994, 236, 1356–1368. [Google Scholar] [CrossRef]
- Buchko, G.W.; Hewitt, S.N.; Napuli, A.J.; Van Voorhis, W.C.; Myler, P.J. Solution-state NMR structure and biophysical characterization of zinc-substituted rubredoxin B (Rv3250c) from Mycobacterium tuberculosis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Blake, P.R.; Park, J.B.; Zhou, Z.H.; Hare, D.R.; Adams, M.W.; Summers, M.F. Solution-state structure by NMR of zinc-substituted rubredoxin from the marine hyperthermophilic archaebacterium Pyrococcus furiosus. Protein Sci. 1992, 1, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Sieker, L.C.; Stenkamp, R.E.; LeGall, J. Rubredoxin in crystalline state. Methods Enzymol. 1994, 243, 203–216. [Google Scholar] [PubMed]
- Dauter, Z.; Wilson, K.S.; Sieker, L.C.; Moulis, J.M.; Meyer, J. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: A high-precision model of a ZnS4 coordination unit in a protein. Proc. Natl. Acad. Sci. USA 1996, 93, 8836–8840. [Google Scholar] [CrossRef] [PubMed]
- Schweimer, K.; Hoffmann, S.; Wastl, J.; Maier, U.G.; Rosch, P.; Sticht, H. Solution structure of a zinc substituted eukaryotic rubredoxin from the cryptomonad alga Guillardia theta. Protein Sci. 2000, 9, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The STAS domain—A link between anion transporters and antisigma-factor antagonists. Curr. Biol. 2000, 10, R53–R55. [Google Scholar] [CrossRef]
- Sharma, A.K.; Ye, L.; Baer, C.E.; Shanmugasundaram, K.; Alber, T.; Alper, S.L.; Rigby, A.C. Solution structure of the guanine nucleotide-binding STAS domain of SLC26-related SulP protein Rv1739c from Mycobacterium tuberculosis. J. Biol. Chem. 2011, 286, 8534–8544. [Google Scholar] [CrossRef] [PubMed]
- Cavet, J.S.; Meng, W.; Pennella, M.A.; Appelhoff, R.J.; Giedroc, D.P.; Robinson, N.J. A nickel-cobalt-sensing ArsR-SmtB family repressor. Contributions of cytosol and effector binding sites to metal selectivity. J. Biol. Chem. 2002, 277, 38441–38448. [Google Scholar] [CrossRef] [PubMed]
- Busenlehner, L.S.; Pennella, M.A.; Giedroc, D.P. The SmtB/ArsR family of metalloregulatory transcriptional repressors: Structural insights into prokaryotic metal resistance. FEMS Microbiol. Rev. 2003, 27, 131–143. [Google Scholar] [CrossRef]
- Osman, D.; Cavet, J.S. Bacterial metal-sensing proteins exemplified by ArsR-SmtB family repressors. Nat. Prod. Rep. 2010, 27, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Ciofi-Baffoni, S.; Cavet, J.S.; Dennison, C.; Graham, A.I.; Harvie, D.R.; Robinson, N.J. NMR structural analysis of cadmium sensing by winged helix repressor CmtR. J. Biol. Chem. 2007, 282, 30181–30188. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Chakravorty, D.K.; Chang, F.M.; Reyes-Caballero, H.; Ye, Y.; Merz, K.M., Jr.; Giedroc, D.P. Solution structure of Mycobacterium tuberculosis NmtR in the apo state: Insights into Ni(II)-mediated allostery. Biochemistry 2012, 51, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.W.; Shankar, S.; Filter, J.J. RNA polymerase elongation factors. Annu. Rev. Microbiol. 2008, 62, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; Schweimer, K.; Burmann, B.M.; Richter, A.; Guttler, S.; Wohrl, B.M.; Rosch, P. The two domains of Mycobacterium tuberculosis NusG protein are dynamically independent. J. Biomol. Struct. Dyn. 2016, 34, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 2016, 80, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Muskett, F.W.; Waters, L.C.; Addis, P.W.; Rieck, B.; Munder, T.; Schleier, S.; Forti, F.; Ghisotti, D.; Carr, M.D.; et al. Mycobacterium tuberculosis RNA polymerase-binding protein A (RbpA) and its interactions with sigma factors. J. Biol. Chem. 2013, 288, 14438–14450. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, R.; Provvedi, R.; Rodrigue, S.; Beaucher, J.; Gaudreau, L.; Smith, I. Sigma factors and global gene regulation in Mycobacterium tuberculosis. J. Bacteriol. 2004, 186, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Morichaud, Z.; Chen, S.; Leonetti, J.P.; Brodolin, K. Mycobacterium tuberculosis RbpA protein is a new type of transcriptional activator that stabilizes the sigma A-containing RNA polymerase holoenzyme. Nucleic. Acids. Res. 2012, 40, 6547–6557. [Google Scholar] [CrossRef] [PubMed]
- Walters, S.B.; Dubnau, E.; Kolesnikova, I.; Laval, F.; Daffe, M.; Smith, I. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 2006, 60, 312–330. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Asensio, J.; Mostowy, S.; Harders-Westerveen, J.; Huygen, K.; Hernandez-Pando, R.; Thole, J.; Behr, M.; Gicquel, B.; Martin, C. PhoP: A missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS ONE 2008, 3, e3496. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Goyal, R.; Sinha, A.; Sarkar, D. Domain structure of virulence-associated response regulator PhoP of Mycobacterium tuberculosis: Role of the linker region in regulator-promoter interaction(s). J. Biol. Chem. 2010, 285, 34309–34318. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Sarkar, D.; Amer, B.R.; Clubb, R.T. Solution structure of the PhoP DNA-binding domain from Mycobacterium tuberculosis. J. Biomol. NMR 2015, 63, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.R.; Imperial, R.; Wang, L.; Navarre, W.W.; Liu, J. Lsr2 of Mycobacterium represents a novel class of H-NS-like proteins. J. Bacteriol. 2008, 190, 7052–7059. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.R.; Li, Y.; Wang, L.; Sintsova, A.; van Bakel, H.; Tian, S.; Navarre, W.W.; Xia, B.; Liu, J. Lsr2 is a nucleoid-associated protein that targets AT-rich sequences and virulence genes in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 5154–5159. [Google Scholar] [CrossRef] [PubMed]
- Av-Gay, Y.; Everett, M. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 2000, 8, 238–244. [Google Scholar] [CrossRef]
- Koul, A.; Choidas, A.; Treder, M.; Tyagi, A.K.; Drlica, K.; Singh, Y.; Ullrich, A. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.; Duran, R.; Wehenkel, A.; Fernandez, P.; England, P.; Brodin, P.; Cole, S.T.; Zimny-Arndt, U.; Jungblut, P.R.; Cervenansky, C.; et al. Proteomic identification of M. tuberculosis protein kinase substrates: PknB recruits GarA, a FHA domain-containing protein, through activation loop-mediated interactions. J. Mol. Biol. 2005, 350, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, A.E.; Grundner, C.; Echols, N.; Gay, L.M.; Lombana, T.N.; Miecskowski, C.A.; Pullen, K.E.; Sung, P.Y.; Alber, T. Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J. Mol. Microbiol. Biotechnol. 2005, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Alzari, P.M. Towards new tuberculosis drugs. Biochem. Soc. Trans. 2007, 35, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Dyson, P. Evolution of transmembrane protein kinases implicated in coordinating remodeling of gram-positive peptidoglycan: Inside versus outside. J. Bacteriol. 2006, 188, 7470–7476. [Google Scholar] [CrossRef] [PubMed]
- Barthe, P.; Mukamolova, G.V.; Roumestand, C.; Cohen-Gonsaud, M. The structure of PknB extracellular PASTA domain from Mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 2010, 18, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Abbott, D.W.; Park, S.T.; Dascher, C.C.; Cantley, L.C.; Husson, R.N. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: Substrate identification and regulation of cell shape. Genes Dev. 2005, 19, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Kelly, G.; Stach, L.; Li, J.; Westcott, S.; Patel, D.; Hunt, D.M.; Howell, S.; Buxton, R.S.; O’Hare, H.M.; et al. An intramolecular switch regulates phosphoindependent FHA domain interactions in Mycobacterium tuberculosis. Sci. Signal. 2009, 2, ra12. [Google Scholar] [CrossRef] [PubMed]
- Roumestand, C.; Leiba, J.; Galophe, N.; Margeat, E.; Padilla, A.; Bessin, Y.; Barthe, P.; Molle, V.; Cohen-Gonsaud, M. Structural insight into the Mycobacterium tuberculosis Rv0020c protein and its interaction with the PknB kinase. Structure 2011, 19, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Gonsaud, M.; Barthe, P.; Canova, M.J.; Stagier-Simon, C.; Kremer, L.; Roumestand, C.; Molle, V. The Mycobacterium tuberculosis Ser/Thr kinase substrate Rv2175c is a DNA-binding protein regulated by phosphorylation. J. Biol. Chem. 2009, 284, 19290–19300. [Google Scholar] [CrossRef] [PubMed]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Stehle, T.; Sreeramulu, S.; Lohr, F.; Richter, C.; Saxena, K.; Jonker, H.R.; Schwalbe, H. The apo-structure of the low molecular weight protein-tyrosine phosphatase A (MptpA) from Mycobacterium tuberculosis allows for better target-specific drug development. J. Biol. Chem. 2012, 287, 34569–34582. [Google Scholar] [CrossRef] [PubMed]
- Van Rompay, A.R.; Johansson, M.; Karlsson, A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol. Ther. 2000, 87, 189–198. [Google Scholar] [CrossRef]
- Munier-Lehmann, H.; Burlacu-Miron, S.; Craescu, C.T.; Mantsch, H.H.; Schultz, C.P. A new subfamily of short bacterial adenylate kinases with the Mycobacterium tuberculosis enzyme as a model: A predictive and experimental study. Proteins 1999, 36, 238–248. [Google Scholar] [CrossRef]
- Miron, S.; Munier-Lehmann, H.; Craescu, C.T. Structural and dynamic studies on ligand-free adenylate kinase from Mycobacterium tuberculosis revealed a closed conformation that can be related to the reduced catalytic activity. Biochemistry 2004, 43, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Mukamolova, G.V.; Kaprelyants, A.S.; Young, D.I.; Young, M.; Kell, D.B. A bacterial cytokine. Proc. Natl. Acad. Sci. USA 1998, 95, 8916–8921. [Google Scholar] [CrossRef] [PubMed]
- Downing, K.J.; Mischenko, V.V.; Shleeva, M.O.; Young, D.I.; Young, M.; Kaprelyants, A.S.; Apt, A.S.; Mizrahi, V. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 2005, 73, 3038–3043. [Google Scholar] [CrossRef] [PubMed]
- Mukamolova, G.V.; Turapov, O.A.; Young, D.I.; Kaprelyants, A.S.; Kell, D.B.; Young, M. A family of autocrine growth factors in Mycobacterium tuberculosis. Mol. Microbiol. 2002, 46, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Gonsaud, M.; Barthe, P.; Bagneris, C.; Henderson, B.; Ward, J.; Roumestand, C.; Keep, N.H. The structure of a resuscitation-promoting factor domain from Mycobacterium tuberculosis shows homology to lysozymes. Nat. Struct. Mol. Biol. 2005, 12, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Maione, V.; Ruggiero, A.; Russo, L.; De Simone, A.; Pedone, P.V.; Malgieri, G.; Berisio, R.; Isernia, C. NMR Structure and Dynamics of the Resuscitation Promoting Factor RpfC Catalytic Domain. PLoS ONE 2015, 10, e0142807. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.A.; Schneider, T.R.; Sieker, L.C.; Dauter, Z.; Lamzin, V.S.; Wilson, K.S. Refinement of triclinic hen egg-white lysozyme at atomic resolution. Acta Crystallogr. Sect. D 1998, 54, 522–546. [Google Scholar] [CrossRef]
- Bal, N.C.; Agrawal, H.; Meher, A.K.; Arora, A. Characterization of peptidyl-tRNA hydrolase encoded by open reading frame Rv1014c of Mycobacterium tuberculosis H37Rv. Biol. Chem. 2007, 388, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Pulavarti, S.V.; Jain, A.; Pathak, P.P.; Mahmood, A.; Arora, A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 2008, 378, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Alix, E.; Blanc-Potard, A.B. MgtC: A key player in intramacrophage survival. Trends Microbiol. 2007, 15, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Labesse, G.; Carrere-Kremer, S.; Esteves, K.; Kremer, L.; Cohen-Gonsaud, M.; Blanc-Potard, A.B. The C-terminal domain of the virulence factor MgtC is a divergent ACT domain. J. Bacteriol. 2012, 194, 6255–6263. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Akif, M.; Khare, G.; Tyagi, A.K.; Mande, S.C.; Sardesai, A.A. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 2008, 190, 7087–7095. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L.; Neumann, T.S.; Cai, S.; Sem, D.S. Solution structures of Mycobacterium tuberculosis thioredoxin C and models of intact thioredoxin system suggest new approaches to inhibitor and drug design. Proteins 2013, 81, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Newton, G.L.; Ko, M.; Martinez, G.J.; Fahey, R.C.; Av-Gay, Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, K.; Buts, L.; Foloppe, N.; Vertommen, D.; Van Belle, K.; Wahni, K.; Roos, G.; Nilsson, L.; Mateos, L.M.; Rawat, M.; et al. Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol. Microbiol. 2012, 86, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L. Thioredoxin—A fold for all reasons. Structure 1995, 3, 245–250. [Google Scholar] [CrossRef]
- Ratledge, C. Iron, mycobacteria and tuberculosis. Tuberculosis 2004, 84, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.M.; Barry, C.E., 3rd; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Snow, G.A. Mycobactins: Iron-chelating growth factors from mycobacteria. Bacteriol. Rev. 1970, 34, 99–125. [Google Scholar] [PubMed]
- Gobin, J.; Moore, C.H.; Reeve, J.R., Jr.; Wong, D.K.; Gibson, B.W.; Horwitz, M.A. Iron acquisition by Mycobacterium tuberculosis: Isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. USA 1995, 92, 5189–5193. [Google Scholar] [CrossRef] [PubMed]
- Quadri, L.E.; Sello, J.; Keating, T.A.; Weinreb, P.H.; Walsh, C.T. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem. Biol. 1998, 5, 631–645. [Google Scholar] [CrossRef]
- Buchko, G.W.; Kim, C.Y.; Terwilliger, T.C.; Myler, P.J. Solution structure of Rv2377c-founding member of the MbtH-like protein family. Tuberculosis 2010, 90, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.M.; Jones, C.M.; Xi, Z.; Speer, A.; Danilchanka, O.; Doornbos, K.S.; Sun, P.; Wu, F.; Tian, C.; Niederweis, M. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003120. [Google Scholar] [CrossRef] [PubMed]
- Symmons, M.F.; Bokma, E.; Koronakis, E.; Hughes, C.; Koronakis, V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2009, 106, 7173–7178. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, Y.; Shashkina, E.; Johnson, S.; Bifani, P.J.; Kurepina, N.; Kreiswirth, B.; Bhattacharya, S.; Spencer, J.; Rendon, A.; Catanzaro, A.; et al. Immunological characterization of novel secreted antigens of Mycobacterium tuberculosis. Scand. J. Immunol. 2005, 61, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.D.; Bloemink, M.J.; Dentten, E.; Whelan, A.O.; Gordon, S.V.; Kelly, G.; Frenkiel, T.A.; Hewinson, R.G.; Williamson, R.A. Solution structure of the Mycobacterium tuberculosis complex protein MPB70: From tuberculosis pathogenesis to inherited human corneal desease. J. Biol. Chem. 2003, 278, 43736–43743. [Google Scholar] [CrossRef] [PubMed]
- Clout, N.J.; Tisi, D.; Hohenester, E. Novel fold revealed by the structure of a FAS1 domain pair from the insect cell adhesion molecule fasciclin I. Structure 2003, 11, 197–203. [Google Scholar] [CrossRef]
- Wang, Z.; Potter, B.M.; Gray, A.M.; Sacksteder, K.A.; Geisbrecht, B.V.; Laity, J.H. The solution structure of antigen MPT64 from Mycobacterium tuberculosis defines a new family of beta-grasp proteins. J. Mol. Biol. 2007, 366, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.G.; Urs, T.A.; Ranganath, R.R. MPT 64 Antigen detection for Rapid confirmation of M.tuberculosis isolates. BMC Res. Notes 2011, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Arora, J.; Kumar, G.; Verma, A.K.; Bhalla, M.; Sarin, R.; Myneedu, V.P. Utility of MPT64 Antigen Detection for Rapid Confirmation of Mycobacterium tuberculosis Complex. J. Glob. Infect. Dis. 2015, 7, 66–69. [Google Scholar] [PubMed]
- Bitter, W.; Houben, E.N.; Bottai, D.; Brodin, P.; Brown, E.J.; Cox, J.S.; Derbyshire, K.; Fortune, S.M.; Gao, L.Y.; Liu, J.; et al. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 2009, 5, e1000507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, A.M.; Gey van Pittius, N.C.; Champion, P.A.; Cox, J.; Luirink, J.; Vandenbroucke-Grauls, C.M.; Appelmelk, B.J.; Bitter, W. Type VII secretion—Mycobacteria show the way. Nat. Rev. Microbiol. 2007, 5, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Ramakrishnan, L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009, 136, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.M.; Savage, N.D.; van Zon, M.; Wilson, L.; Vandenbroucke-Grauls, C.M.; van der Wel, N.N.; Ottenhoff, T.H.; Bitter, W. The ESX-5 secretion system of Mycobacterium marinum modulates the macrophage response. J. Immunol. 2008, 181, 7166–7175. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, M.S.; Unnikrishnan, M.; McConnell, M.J.; Borowsky, M.; Cheng, T.Y.; Siddiqi, N.; Fortune, S.M.; Moody, D.B.; Rubin, E.J. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc. Natl. Acad. Sci. USA 2009, 106, 18792–18797. [Google Scholar] [CrossRef] [PubMed]
- Arbing, M.A.; Kaufmann, M.; Phan, T.; Chan, S.; Cascio, D.; Eisenberg, D. The crystal structure of the Mycobacterium tuberculosis Rv3019c-Rv3020c ESX complex reveals a domain-swapped heterotetramer. Protein Sci. 2010, 19, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, K.L.; Renshaw, P.S.; Collins, M.L.; Wright, R.L.; Hunt, D.M.; Gordon, S.V.; Hewinson, R.G.; Buxton, R.S.; Williamson, R.A.; Carr, M.D. Characterisation of complex formation between members of the Mycobacterium tuberculosis complex CFP-10/ESAT-6 protein family: Towards an understanding of the rules governing complex formation and thereby functional flexibility. FEMS Microbiol. Lett. 2004, 238, 255–262. [Google Scholar] [PubMed]
- Renshaw, P.S.; Panagiotidou, P.; Whelan, A.; Gordon, S.V.; Hewinson, R.G.; Williamson, R.A.; Carr, M.D. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J. Biol. Chem. 2002, 277, 21598–21603. [Google Scholar] [PubMed]
- Renshaw, P.S.; Lightbody, K.L.; Veverka, V.; Muskett, F.W.; Kelly, G.; Frenkiel, T.A.; Gordon, S.V.; Hewinson, R.G.; Burke, B.; Norman, J.; et al. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 2005, 24, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Ilghari, D.; Lightbody, K.L.; Veverka, V.; Waters, L.C.; Muskett, F.W.; Renshaw, P.S.; Carr, M.D. Solution structure of the Mycobacterium tuberculosis EsxG.EsxH complex: Functional implications and comparisons with other M. tuberculosis Esx family complexes. J. Biol. Chem. 2011, 286, 29993–30002. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, C.; Papavinasasundaram, K.G.; Speight, R.A.; Springer, B.; Sander, P.; Bottger, E.C.; Colston, M.J.; Draper, P. The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol. Microbiol. 2002, 46, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Molle, V.; Saint, N.; Campagna, S.; Kremer, L.; Lea, E.; Draper, P.; Molle, G. pH-dependent pore-forming activity of OmpATb from Mycobacterium tuberculosis and characterization of the channel by peptidic dissection. Mol. Microbiol. 2006, 61, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Huff, J.; Janik, K.; Walter, K.; Keller, C.; Ehlers, S.; Bossmann, S.H.; Niederweis, M. Expression of the ompATb operon accelerates ammonia secretion and adaptation of Mycobacterium tuberculosis to acidic environments. Mol. Microbiol. 2011, 80, 900–918. [Google Scholar] [CrossRef] [PubMed]
- Teriete, P.; Yao, Y.; Kolodzik, A.; Yu, J.; Song, H.; Niederweis, M.; Marassi, F.M. Mycobacterium tuberculosis Rv0899 adopts a mixed alpha/beta-structure and does not form a transmembrane beta-barrel. Biochemistry 2010, 49, 2768–2777. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Auguin, D.; Delbecq, S.; Dumas, E.; Molle, G.; Molle, V.; Roumestand, C.; Saint, N. Structure of the Mycobacterium tuberculosis OmpATb protein: A model of an oligomeric channel in the mycobacterial cell wall. Proteins 2011, 79, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, C.; Gao, Y.; Wu, K.; Shi, P.; Lai, C.; Chen, L.; Wu, F.; Tian, C. Structural studies of Mycobacterium tuberculosis Rv0899 reveal a monomeric membrane-anchoring protein with two separate domains. J. Mol. Biol. 2012, 415, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Barghava, N.; Kim, J.; Niederweis, M.; Marassi, F.M. Molecular structure and peptidoglycan recognition of Mycobacterium tuberculosis ArfA (Rv0899). J. Mol. Biol. 2012, 416, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Yeats, C.; Bateman, A. The BON domain: A putative membrane-binding domain. Trends Biochem. Sci. 2003, 28, 352–355. [Google Scholar] [CrossRef]
- Pautsch, A.; Schulz, G.E. High-resolution structure of the OmpA membrane domain. J. Mol. Biol. 2000, 298, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Buchko, G.W.; Kim, C.Y.; Terwilliger, T.C.; Kennedy, M.A. Solution structure of the conserved hypothetical protein Rv2302 from Mycobacterium tuberculosis. J. Bacteriol. 2006, 188, 5993–6001. [Google Scholar] [CrossRef] [PubMed]
- Buchko, G.W.; Phan, I.; Myler, P.J.; Terwilliger, T.C.; Kim, C.Y. Inaugural structure from the DUF3349 superfamily of proteins, Mycobacterium tuberculosis Rv0543c. Arch. Biochem. Biophys. 2011, 506, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Rath, P.; Huang, C.; Wang, T.; Wang, T.; Li, H.; Prados-Rosales, R.; Elemento, O.; Casadevall, A.; Nathan, C.F. Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E4790–E4797. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, E.; Kerff, F.; Terrak, M.; Ayala, J.A.; Charlier, P. The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvanese, L.; Falcigno, L.; Maglione, C.; Marasco, D.; Ruggiero, A.; Squeglia, F.; Berisio, R.; D’Auria, G. Structural and binding properties of the PASTA domain of PonA2, a key penicillin binding protein from Mycobacterium tuberculosis. Biopolymers 2014, 101, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Barthe, P.; Veyron-Churlet, R.; de Visch, A.; Gilleron, M.; Saliou, J.M.; Tomavo, S.; Nigou, J.; Brodin, P.; Cohen-Gonsaud, M. Mycobacterium tuberculosis LppM Displays an Original Structure and Domain Composition Linked to a Dual Localization. Structure 2016, 24, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Poquet, Y.; Levillain, F.; Peguillet, I.; Larrouy-Maumus, G.; Gilleron, M.; Ewann, F.; Christophe, T.; Fenistein, D.; Jang, J.; et al. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. 2010, 6, e1001100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, B.; Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Int. Ed. Engl. 2003, 42, 864–890. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Bach, H.; Wong, D.; Av-Gay, Y. Mycobacterium tuberculosis PtkA is a novel protein tyrosine kinase whose substrate is PtpA. Biochem. J. 2009, 420, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Dalvit, C.; Pevarello, P.; Tato, M.; Veronesi, M.; Vulpetti, A.; Sundstrom, M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol. NMR 2000, 18, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.F.; Johnson, C.S., Jr. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Nassau, P.M.; Martin, S.L.; Brown, R.E.; Weston, A.; Monsey, D.; McNeil, M.R.; Duncan, K. Galactofuranose biosynthesis in Escherichia coli K-12: Identification and cloning of UDP-galactopyranose mutase. J. Bacteriol. 1996, 178, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Jackson, M.; Ma, Y.; McNeil, M. Cell wall core galactofuran synthesis is essential for growth of mycobacteria. J. Bacteriol. 2001, 183, 3991–3998. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Shi, Y.; Colombo, C.; Kuttiyatveetil, J.R.; Zalatar, N.; van Straaten, K.E.; Mohan, S.; Sanders, D.A.; Pinto, B.M. A Second, Druggable Binding Site in UDP-Galactopyranose Mutase from Mycobacterium tuberculosis? Chembiochem 2016, 17, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, H.; Johnston, J.B.; Ortiz de Montellano, P.R. The Mycobacterium tuberculosis cytochrome P450 system. Arch. Biochem. Biophys. 2010, 493, 82–95. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.J.; Belcher, J.; Driscoll, M.D.; Fernandez, C.C.; Le Van, D.; Bui, S.; Golovanova, M.; Munro, A.W. The Mycobacterium tuberculosis cytochromes P450: Physiology, biochemistry & molecular intervention. Future Med. Chem. 2010, 2, 1339–1353. [Google Scholar] [PubMed]
- Hudson, S.A.; McLean, K.J.; Munro, A.W.; Abell, C. Mycobacterium tuberculosis cytochrome P450 enzymes: A cohort of novel TB drug targets. Biochem. Soc. Trans. 2012, 40, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Belin, P.; Le Du, M.H.; Fielding, A.; Lequin, O.; Jacquet, M.; Charbonnier, J.B.; Lecoq, A.; Thai, R.; Courcon, M.; Masson, C.; et al. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7426–7431. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.J.; Carroll, P.; Lewis, D.G.; Dunford, A.J.; Seward, H.E.; Neeli, R.; Cheesman, M.R.; Marsollier, L.; Douglas, P.; Smith, W.E.; et al. Characterization of active site structure in CYP121. A cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 2008, 283, 33406–33416. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: Mechanisms of action. Trends Microbiol. 2003, 11, 272–279. [Google Scholar] [CrossRef]
- Hudson, S.A.; McLean, K.J.; Surade, S.; Yang, Y.Q.; Leys, D.; Ciulli, A.; Munro, A.W.; Abell, C. Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem. Int. Ed. Engl. 2012, 51, 9311–9316. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Classification | Protein | Brief Description | PDB ID (Deposit Year) |
|---|---|---|---|
| Transport-related proteins | Rv2244 | Acyl carrier protein | 1KLP (2001) |
| Rv3250c * | Electron transport | 2KN9 (2009) | |
| Rv1739c | Sulfate transporter | 2KLN (2009) | |
| Transcription-related proteins | MT3852 | ArsR family, transcription regulator, nickel metal sensor | 2LKP (2012) |
| Rv1994c | ArsR family, transcription regulator, cadmium metal sensor | 2JSC (2013) | |
| Rv0639 * | Transcription elongation–termination factor | 2MI6 (2013) | |
| Rv2050 * | RNA polymerase binding protein | 2M4V (2013) | |
| Nucleotide-binding proteins | J113_05350 | DNA-binding response regulator | 2RV8 (2015) |
| Rv3597c * | Nucleoid-associated protein | 2KNG (2009) | |
| Ser/Thr Protein kinase-related proteins | Rv0014c * | Ser/Thr protein kinase (STPK) PknB | 2KUD, 2KUE, 2KUF, 2KUI (2010) |
| Rv1827 | Substrate of STPK PknB | 2KFU (2009) | |
| Rv0020c | Substrate of STPK PknB | 2LC0, 2LC1 (2011) | |
| Rv2175c | Substrate of STPK PknL | 2KFS (2009) | |
| Rv2234 * | Protein-tyrosine Phosphatase | 2LUO (2012) | |
| Enzymes and related proteins | Rv0733 * | Transferase, adenylate kinase | 1P4S (2003) |
| Rv1009 | Hydrolase, Resuscitation-promoting factor | 1XSF (2004) | |
| Rv1884c | Hydrolase, Resuscitation-promoting factor | 2N5Z (2015) | |
| Rv1014c * | Hydrolase, peptidyl-tRNA hydrolase | 2JRC (2007) | |
| Rv2737c | Hydrolase, endonuclease | 2L8L (2011) | |
| MT1859 | Hydrolase | 2LQJ (2012) | |
| Rv3914 | Thioredoxin | 2L59, 2L4Q (2010) | |
| Rv3198.1 | Mycothiol-dependent reductase | 2LQQ, 2LQO (2012) | |
| Siderophore-related proteins | Rv2377c * | Siderophore biosynthesis | 2KHR (2009) |
| Rv0451c | Siderophore export | 2LW3 (2012) | |
| Secreted proteins | Rv2875 | Immunogenic protein MPT70 | 1NYO (2003) |
| Rv1980c | Immunogenic protein MPT64 | 2HHI (2006) | |
| Rv3875/Mb3904 | Type VII secretion system protein | 1WA8 (2004) | |
| Rv0287/Rv0288 | Type VII secretion system protein | 2KG7 (2009) | |
| Rv0603 | Immune system | 2KGY (2009) | |
| 2LRA (2012) | |||
| Membrane proteins | Rv1761c | Membrane protein | 2K3M (2008) |
| Rv0899 | Pore-forming outer membrane protein | 2KGS, 2KGW (2009) | |
| 2KSM (2010) | |||
| 2L26 (2010) | |||
| 2LBT, 2LCA (2011) | |||
| Uncharacterized proteins | Rv2302 | Unknown function | 2A7Y (2005) |
| Rv0543c | Domain of Unknown Function DUF3349 (PF11829) | 2KVC (2010) | |
| Other proteins | Rv3418c | Chaperone | 1P82, 1P83 (2003) |
| Rv1311 | ATP synthase epsilon chain | 2LX5 (2012) | |
| Rv0431 | Vesiculogenesis and immune response regulation | 2M5Y (2013) | |
| Rv3682 * | Penicillin binding protein | 2MGV (2013) | |
| Rv1466 | [Fe-S] cluster assembly related protein | 5IRD (2016) | |
| Rv2171 | Lipoprotein | 2NC8 (2016) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-H.; Kang, S.-M.; Lee, B.-J. Solution NMR Studies of Mycobacterium tuberculosis Proteins for Antibiotic Target Discovery. Molecules 2017, 22, 1447. https://doi.org/10.3390/molecules22091447
Kim D-H, Kang S-M, Lee B-J. Solution NMR Studies of Mycobacterium tuberculosis Proteins for Antibiotic Target Discovery. Molecules. 2017; 22(9):1447. https://doi.org/10.3390/molecules22091447
Chicago/Turabian StyleKim, Do-Hee, Sung-Min Kang, and Bong-Jin Lee. 2017. "Solution NMR Studies of Mycobacterium tuberculosis Proteins for Antibiotic Target Discovery" Molecules 22, no. 9: 1447. https://doi.org/10.3390/molecules22091447
APA StyleKim, D.-H., Kang, S.-M., & Lee, B.-J. (2017). Solution NMR Studies of Mycobacterium tuberculosis Proteins for Antibiotic Target Discovery. Molecules, 22(9), 1447. https://doi.org/10.3390/molecules22091447