3.2. Procedures
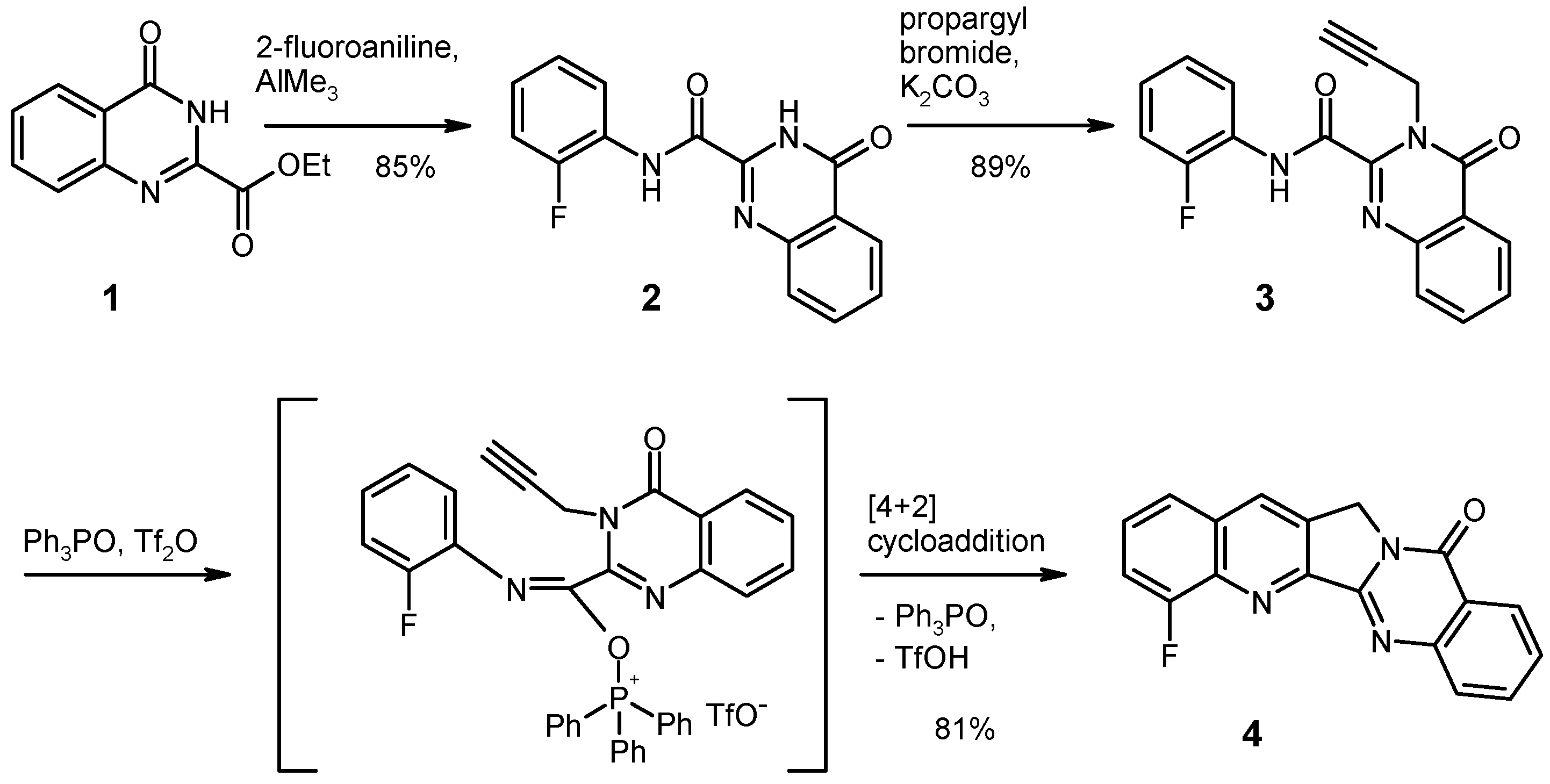
N-(2-Fluorophenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (2). To a solution of 2-fluoroaniline (0.889 g, 8 mmol) in dry 1,2-dichloroethane (20 mL) under argon was added drop wise a 2 M solution of AlMe3 (4.0 mL, 8 mmol) in heptane. The mixture was stirred for 30 min at room temperature, then the ester 1 (1.091 g, 5 mmol) was added in one portion, and the mixture was refluxed for 2 h. After cooling to 0 °C, it was then quenched by slow addition of 2 N HCl (20 mL), followed by water (80 mL). The mixture was exhaustively extracted with CH2Cl2 and the combined extracts were washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was recrystallized from EtOH to give 2 (1.202 g, 85%) as colorless needles, m.p. 216–217 °C. MS (EI, 70 eV) m/z = 283 (M+, 32%), 236 (18), 146 (91), 119 (100), 118 (18), 91 (15), 90 (31), 83 (12); 1H-NMR (DMSO-d6) δ: 12.62 (br s, 1H, NH), 10.50 (br s, 1H, NH), 8.21 (dd, J = 8.0 Hz, 1.2 Hz, 1H, 5-H), 7.97–7.89 (m, 2H, 7-H, phenyl 6′-H), 7.87 (dd, J = 8.2 Hz, 0.9 Hz, 1H, 8-H), 7.65 (ddd, J = 8.2 Hz, 7.0 Hz, 1.3 Hz, 1H, 6-H), 7.41–7.23 (m, 3H, phenyl 3′-H, 4′-H, 5′-H); 13C-NMR (DMSO-d6) δ: 161.3, 158.0, 154.5 (d, JC–F = 246.9 Hz), 146.6, 145.6, 134.8, 128.3, 127.7, 126.9 (d, JC–F = 7.7 Hz), 126.2, 124.8, 124.7, 124.6, 122.8, 115.8 (d, JC–F = 19.4 Hz). Anal. calcd. for C15H10FN3O2·0.2 H2O: C, 62.80; H, 3.65; N, 14.65. Found: C, 62.79; H, 3.46; N, 14.66. HRMS (ESI-TOF) m/z calcd. for C15H11FN3O2 ([M + H]+): 284.0830. Found: 284.0827.
N-(2-Fluorophenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (3). To a solution of the anilide 2 (0.566 g, 2 mmol) in dry DMF (15 mL) were added K2CO3 (0.304 g, 2.2 mmol) and propargyl bromide (0.327 g of an 80% solution in toluene, 2.2 mmol). The mixture was stirred at room temperature for 24 h, then it was poured into water (100 mL) and it was extracted with CH2Cl2 (3 × 100 mL). The combined extracts were washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was recrystallized from EtOH to give 3 (0.527 g, 89%) as colorless crystals, m.p. 153–154 °C. MS (EI, 70 eV) m/z = 321 (M+, 21%), 320 (72), 301 (27), 292 (52), 184 (25), 155 (54), 148 (40), 145 (36), 129 (100), 119 (96), 90 (64), 83 (47), 75 (42), 63 (41); 1H-NMR (CDCl3) δ: 9.99 (s, 1H, NH), 8.52–8.41 (m, 1H, phenyl 6′-H), 8.37 (ddd, J = 8.0 Hz, 1.3 Hz, 0.6 Hz, 1H, 5-H), 7.89–7.79 (m, 2H, 7-H, 8-H), 7.62 (ddd, J = 8.2 Hz, 6.5 Hz, 1.9 Hz, 1H, 6-H), 7.24–7.14 (m, 3H, phenyl 3′-H, 4′-H, 5′-H), 5.60 (d, J = 2.5 Hz, 2H, CH2), 2.27 (t, J = 2.5 Hz, 1H, acetylene-H); 13C-NMR (CDCl3) δ: 161.4, 158.3, 153.1 (d, JC–F = 244.0 Hz), 145.0, 144.9, 135.1, 129.3, 128.2, 127.6, 125.7, 125.6 (d, JC–F = 7.5 Hz), 124.9 (d, JC–F = 3.8 Hz), 121.9, 121.7, 115.3 (d, JC–F = 19.1 Hz), 78.9, 72.2, 33.8. Anal. calcd. for C18H12FN3O2·0.2 H2O: C, 66.54; H, 3.85; N, 12.93. Found: C, 66.53; H, 3.64; N, 12.97. HRMS (ESI-TOF) m/z calcd. for C18H13FN3O2 ([M + H]+): 322.0986. Found: 322.0988.
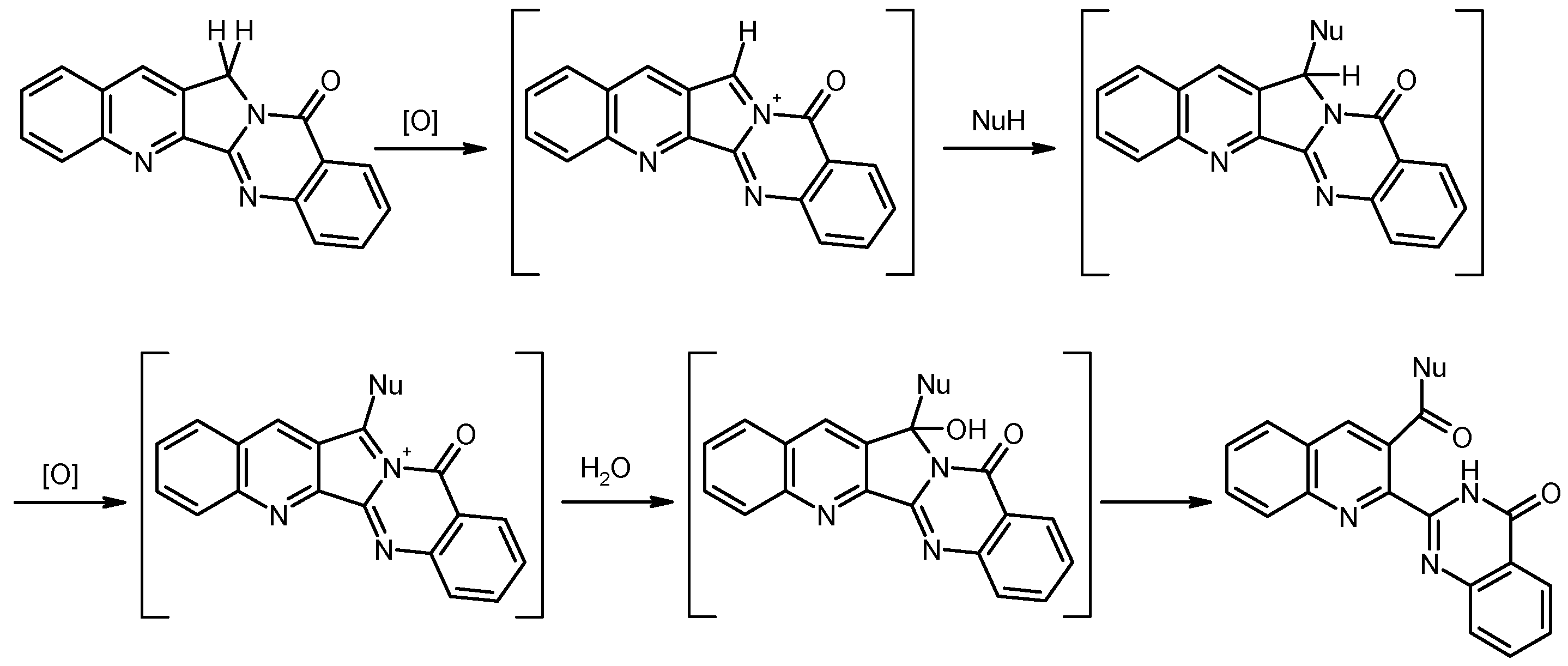
4-Fluoroquinolino[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (4). To a solution of triphenylphosphine oxide (0.835 g, 3 mmol) in dry CH2Cl2 (22 mL) was drop wise added trifluoromethanesulfonic anhydride (0.25 mL, 1.5 mmol) at 0 °C under argon, and the mixture was stirred at the same temperature for 15 min. Then, compound 3 (0.321 g, 1 mmol) was added in one portion at 0 °C, and the mixture was stirred for 1 h while slowly warming to room temperature. The reaction was quenched by addition of 10% aqueous NaHCO3 (15 mL). The phases were separated and the aqueous layer was exhaustively extracted with CH2Cl2. The combined organic layers were washed with water and brine, dried over Na2SO4 and evaporated. To remove excess triphenylphosphine oxide, the residue was subjected to column chromatography, eluting first with CH2Cl2, then with CH2Cl2/ethyl acetate (19:1). The fraction showing an intense blue fluorescence under UV366 was evaporated under reduced pressure and the residue was recrystallized from EtOH to give 4 (0.244 g, 81%) as colorless needles, m.p. > 310 °C (sublimation). MS (EI, 70 eV) m/z = 304 ([M + 1]+, 20%), 303 (M+, 100), 302 (36), 275 (13), 274 (10), 262 (8), 152 (12), 77 (7); 1H-NMR (CDCl3) δ: 8.49 (d, J = 1.0 Hz, 1H, 14-H), 8.42 (dd, J = 8.0 Hz, 1.1 Hz, 1H, 10-H), 8.13–8.08 (m, 1H, 7-H), 7.86 (ddd, J = 8.3 Hz, 7.2 Hz, 1.6 Hz, 1H, 8-H), 7.75 (d, J = 8.3 Hz, 1H, 1-H), 7.68–7.49 (m, 3H, 2-H, 3-H, 9-H), 5.37 (d, J = 0.8 Hz, 2H, CH2); 13C-NMR (CDCl3) δ: 160.7 (11-C), 158.8 (d, JC–F = 261.4 Hz, 4-C), 152.1 (5b-C), 151.7 (5a-C), 149.4 (6a-C), 140.1 (d, JC–F = 12.8 Hz, 4a-C), 134.8 (8-C), 131.6 (d, JC–F = 2.9 Hz, 14-C), 130.7 (13a-C), 130.3 (14a-C), 129.1 (7-C), 128.7 (d, JC–F = 7.8 Hz, 2-C), 127.8 (9-C), 126.6 (10-C), 123.7 (d, JC–F = 5.0 Hz, 1-C), 121.5 (10a-C), 114.9 (d, JC–F = 18.7 Hz, 3-C), 47.4 (13-C). Anal. calcd. for C18H10FN3O·0.15 H2O: C, 70.65; H, 3.39; N, 13.73. Found: C, 70.63; H, 3.30; N, 13.74. HRMS (ESI-TOF) m/z calcd. for C18H11FN3O ([M + H]+): 304.0881. Found: 304.0882.
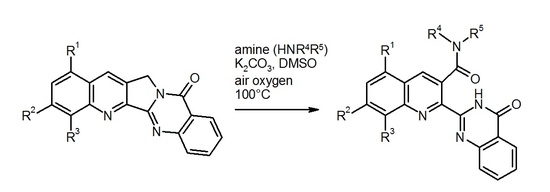
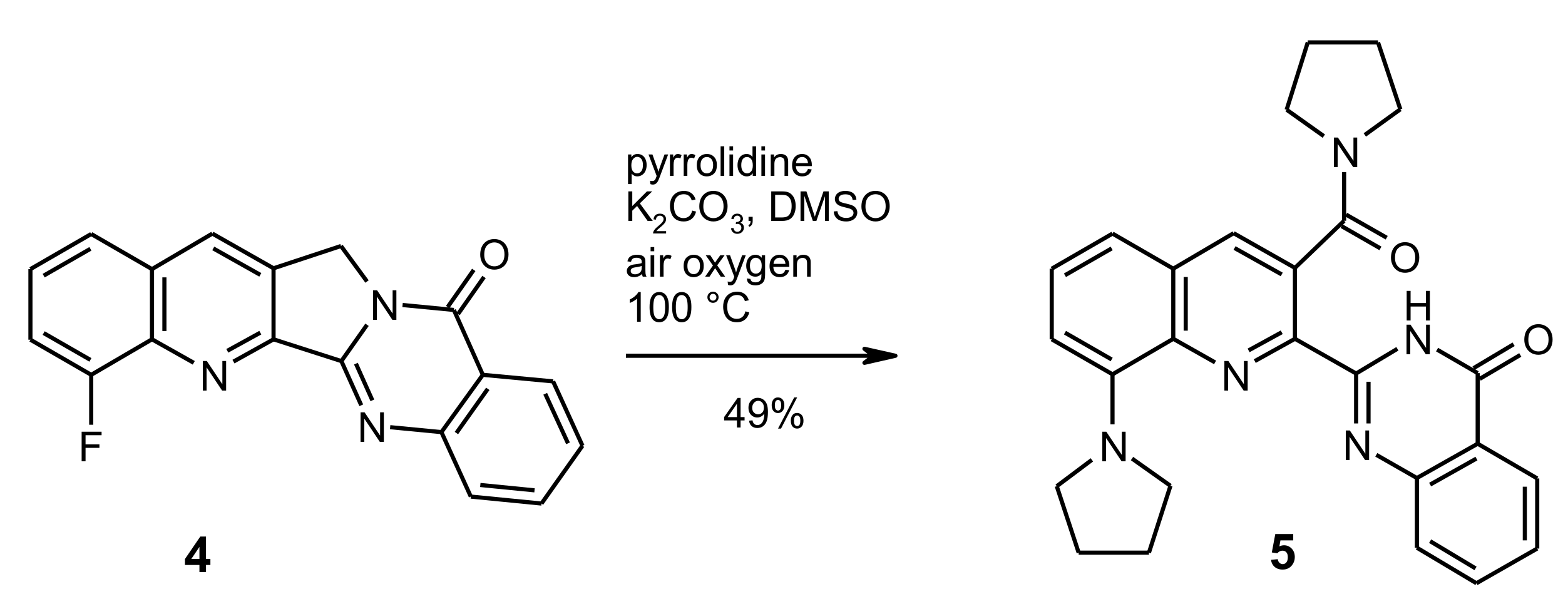
2-[8-(Pyrrolidin-1-yl)-3-(pyrrolidin-1-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (5). To a solution/suspension of the fluoro compound 4 (0.152 g, 0.5 mmol) and K2CO3 (0.069 g, 0.5 mmol) in DMSO (15 mL) was added pyrrolidine (0.356 g, 5 mmol). The mixture was heated to 100 °C and the color of the solution changed into dark red. Stirring was continued for 24 h at 100 °C, then the mixture was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (94:6) to afford compound 5 (108 mg, 49%) as red crystals, m.p. 210–211 °C (CHCl3). MS (EI, 70 eV) m/z = 439 (M+, 9%), 368 (100), 340 (42), 339 (27), 311 (28), 299 (23), 273 (23); 1H-NMR (CDCl3) δ 10.68 (s, 1H, NH), 8.35 (dd, J = 7.9 Hz, 1.1 Hz, 1H, quinazoline 5-H), 8.10 (s, 1H, quinoline 4-H), 7.80–7.73 (m, 1H, quinazoline 7-H), 7.67–7.60 (m, 1H, quinazoline 8-H), 7.55–7.45 (m, 2H, quinazoline 6-H, quinoline 6-H), 7.15 (d, J = 7.2 Hz, 1H, quinoline 5-H), 6.84 (d, J = 7.8 Hz, 1H, quinoline 7-H), 4.00–3.80 (m, 6H, N-arylpyrrolidine 2-H, 2′-H, 5-H, 5′-H, N-acylpyrrolidine 2-H, 2′-H), 3.24 (t, J = 6.7 Hz, 2H, N-acylpyrrolidine 5-H, 5′-H), 2.17–2.10 (m, 4H, N-arylpyrrolidine 3-H, 3′-H, 4-H, 4′-H), 2.06 (quint, J = 6.9 Hz, 2H, N-acylpyrrolidine 3-H, 3′-H), 1.88 (quint, J = 6.8 Hz, 2H, N-acylpyrrolidine 4-H, 4′-H); 13C-NMR (CDCl3) δ: 168.6 (amide C=O), 161.4 (quinazoline 4-C), 149.0 (quinazoline 8a-C), 148.7 (quinazoline 2-C), 146.6 (quinoline 8-C), 139.3 quinoline 2-C), 138.4 (quinoline 8a-C), 136.1 (quinoline 4-C), 134.7 (quinazoline 7-C), 130.8 (quinoline 4a-C), 130.5 (quinoline 3-C), 130.3 (quinoline 6-C), 128.3 (quinazoline 8-C), 127.6 (quinazoline 6-C), 127.0 (quinazoline 5-C), 122.6 (quinazoline 4a-C), 114.9 (quinoline 5-C), 111.3 (quinoline 7-C), 52.6 (N-arylpyrrolidine 2-C, 5-C), 48.6 (N-acylpyrrolidine 5-C), 46.2 (N-acylpyrrolidine 2-C), 26.2 (N-arylpyrrolidine 3-C, 4-C), 26.1 (N-acylpyrrolidine 4-C), 25.0 (N-acylpyrrolidine 3-C). HRMS (ESI-TOF) m/z calcd. for C26H26N5O2 ([M + H]+): 440.2081. Found: 440.2080.
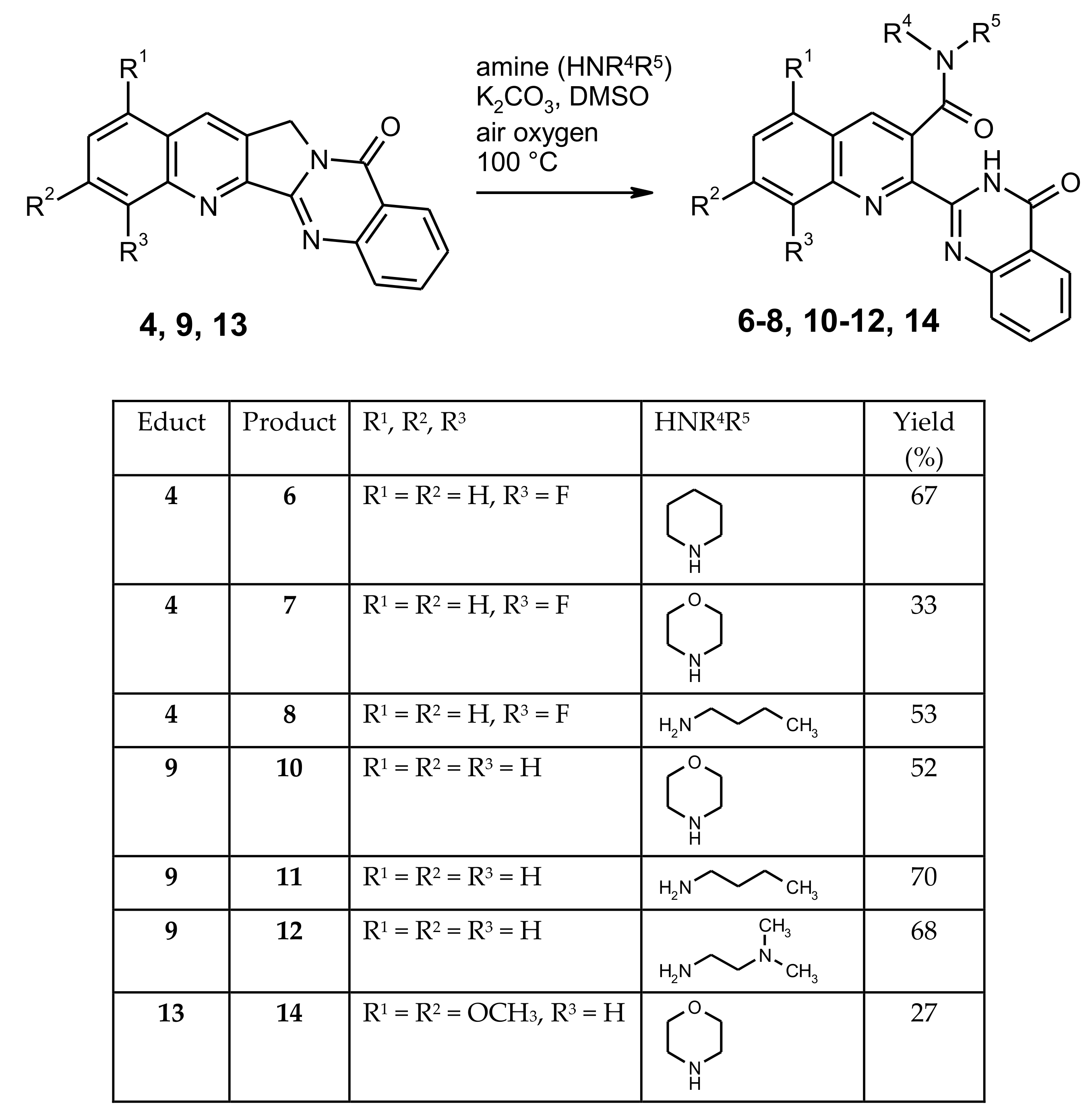
2-[8-Fluoro-3-(piperidin-1-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (6). A mixture of the fluoro compound 4 (0.152 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and piperidine (0.426 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (97:3) to afford 6 (0.134 g, 67%) as dark-red crystals, m.p. 215–216 °C (CHCl3). MS (EI, 70 eV) m/z = 402 (M+, 2%), 318 (26), 289 (16), 171 (21), 119 (36), 84 (100), 69 (26), 64 (21), 57 (41), 56 (44), 55 (67); 1H-NMR (CDCl3) δ 11.11 (s, 1H, NH), 8.38 (ddd, J = 7.9 Hz, 1.5 Hz, 0.5 Hz, 1H, quinazoline 5-H), 8.24 (d, J = 1.4 Hz, 1H, quinoline 4-H), 7.81 (ddd, J = 8.4 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.76 (ddd, J = 8.2 Hz, 1.4 Hz, 0.5 Hz, 1H, quinazoline 8-H), 7.70 (dd, J = 8.2 Hz, 1.2 Hz, 1H, quinoline 5-H), 7.64 (td, J = 7.9 Hz, 4.7 Hz, 1H, quinoline 6-H), 7.59–7.49 (m, 2H, quinazoline 6-H, quinoline 7-H), 4.44–4.27 (m, 1H, piperidine 2-H), 3.50 (ddd, J = 13.3 Hz, 9.8 Hz, 3.6 Hz, 1H, piperidine 2′-H), 3.32–3.22 (m, 1H, piperidine 6-H), 3.12 (ddd, J = 13.1 Hz, 9.6 Hz, 3.4 Hz, 1H, piperidine 6′-H), 2.01–1.87 (m, 1H, piperidine 3-H), 1.84–1.69 (m, 2H, piperidine 3′-H, 4-H), 1.62–1.50 (m, 2H, piperidine 4′-H, 5-H), 1.48–1.41 (m, 1H, piperidine 5′-H); 13C-NMR (CDCl3) δ: 168.2 (amide C=O), 161.4, (quinazoline 4-C), 158.1 (d, JC–F = 261.3 Hz, quinoline 8-C), 148.6 (quinazoline 8a-C), 147.9 (quinazoline 2-C), 144.7 (quinoline 2-C), 136.8 (d, JC–F = 12.4 Hz, quinoline 8a-C), 136.0 (d, JC–F = 2.9 Hz, quinoline 4-C), 134.7 (quinazoline 7-C), 131.4 (quinoline 3-C), 130.0 (quinoline 4a-C), 129.3 (d, JC–F = 7.8 Hz, quinoline 6-C), 128.5 (quinazoline 8-C), 128.2 (quinazoline 6-C), 127.1 (quinazoline 5-C), 123.4 (d, JC–F = 5.0 Hz, quinoline 5-C), 123.0 (quinazoline 4a-C), 115.3 (d, JC–F = 18.3 Hz, quinoline 7-C), 48.2 (piperidine 6-C), 43.0 (piperidine 2-C), 25.8 (piperidine 5-C), 25.4 (piperidine 3-C), 24.7 (piperidine 4-C). HRMS (ESI-TOF) m/z calcd. for C23H20FN4O2 ([M + H]+) 403.1563. Found 403.1563.
2-[8-Fluoro-3-(morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (7). A mixture of the fluoro compound 4 (0.152 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and morpholine (0.436 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to column chromatography, eluting with CH2Cl2/MeOH (97:3) to afford 7 (0.067 g, 33%) as yellow crystals, m.p. > 265 °C (sublimation; CHCl3). MS (EI, 70 eV) m/z = 405 ([M + 1]+, 2%), 404 (M+, 8), 319 (85), 318 (100), 146 (20), 119 (43), 57 (22), 56 (23); 1H-NMR (CDCl3) δ 11.10 (s, 1H, NH), 8.41–8.36 (m, 1H, quinazoline 5-H), 8.28 (d, J = 1.4 Hz, 1H, quinoline 4-H), 7.82 (ddd, J = 8.4 Hz, 7.0 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.77 (ddd, J = 8.2 Hz, 1.4 Hz, 0.6 Hz, 1H, quinazoline 8-H), 7.72 (dd, J = 8.3 Hz, 1.3 Hz, 1H, quinoline 5-H), 7.66 (td, J = 7.9 Hz, 4.6 Hz, 1H, quinoline 6-H), 7.61–7.56 (m, 1H, quinazoline 6-H), 7.56–7.51 (m, 1H, quinoline 7-H), 4.29 (dt, J = 13.0 Hz, 3.0 Hz, 1H, morpholine 5-H), 4.11 (dt, J = 11.7 Hz, 3.6 Hz, 1H, morpholine 6-H), 3.81 (ddd, J = 11.7 Hz, 9.3 Hz, 2.9 Hz, 1H, morpholine 6′-H), 3.74–3.64 (m, 2H, morpholine 2-H, 5′-H), 3.55 (ddd, J = 11.6 Hz, 9.0 Hz, 3.1 Hz, 1H, morpholine 2′-H), 3.38–3.27 (m, 1H, morpholine 3-H), 3.26–3.17 (m, 1H, morpholine 3′-H); 13C-NMR (CDCl3) δ: 168.8 (amide C=O), 161.3 (quinazoline 4-C), 158.0 (JC–F = 260.0 Hz, quinoline 8-C), 148.3 (quinazoline 8a-C), 147.8 (quinazoline 2-C), 144.7 (quinoline 2-C), 136.9 (JC–F = 12.5 Hz, quinoline 8a-C), 136.4 (JC–F = 2.8 Hz, quinoline 4-C), 134.9 (quinazoline 7-C), 130.2 (quinoline 3-C), 129.8 (quinoline 4a-C), 129.6 (JC–F = 7.9 Hz, quinoline 6-C), 128.5 (quinazoline 8-C), 128.4 (quinazoline 6-C), 127.1 (quinazoline 5-C), 123.5 (JC–F = 4.9 Hz, quinoline 5-C), 122.9 (quinazoline 4a-C), 115.6 (JC–F = 18.4 Hz, quinoline 7-C), 66.6 (morpholine 2-C), 66.2 (morpholine 6-C), 47.2 (morpholine 3-C), 42.6 (morpholine 5-C). HRMS (ESI-TOF) m/z calcd. for C22H18FN4O3 ([M + H]+) 405.1357. Found 405.1356.
N-Butyl-8-fluoro-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (8). A mixture of the fluoro compound 4 (0.100 g, 0.33 mmol), K2CO3 (0.046 g, 0.33 mmol) and n-butylamine (0.241 g, 3.3 mmol) in DMSO (10 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/MeOH (96:4) to afford 8 (0.078 g, 53%) as colorless crystals, m.p. 218–220 °C (methyl tert-butyl ether). MS (EI, 70 eV) m/z = 391 ([M + 1]+, 2%), 390 (M+, 6), 319 (63), 318 (100), 291 (28), 171 (20), 146 (21), 119 (49), 92 (22), 90 (35); 1H-NMR (CDCl3) δ 10.94 (s, 1H, quinazolinone NH), 8.39 (d, J = 1.3 Hz, 1H, quinoline 4-H), 8.22 (d, J = 7.9 Hz, 1H, quinazoline 5-H), 7.76–7.74 (m, 2H, quinazoline 7-H, 8-H), 7.68 (d, J = 7.6 Hz, 1H, quinoline 5-H), 7.62 (td, J = 7.9 Hz, 4.8 Hz, 1H, quinoline 6-H), 7.53–7.46 (m, 1H, quinoline 7-H), 7.46–7.38 (m, 1H, quinazoline 6-H), 6.54 (t, J = 5.6 Hz, 1H, amide NH), 3.71–3.58 (m, 2H, butyl 1-CH2), 1.76 (p, J = 7.5 Hz, 2H, butyl 2-CH2), 1.51 (dq, J = 14.6 Hz, 7.3 Hz, 2H, butyl 3-CH2), 1.01 (t, J = 7.4 Hz, 3H, butyl CH3); 13C-NMR (CDCl3) δ: 168.1 (amide C=O), 161.2 (quianzoline 4-C), 157.7 (d, JC–F = 261.3 Hz, quinoline 8-C), 148.2 (quinazoline 8a-C), 147.6 (quinazoline 2-C), 144.6 (quinoline 2-C), 137.6 (d, JC–F = 2.5 Hz, quinoline 4-C), 136.6 (d, JC–F = 12.4 Hz, quinoline 8a-C), 134.6 (quinazoline 7-C), 131.5 (quinoline 3-C), 129.4 (quinoline 4a-C), 129.2 (d, JC–F = 7.9 Hz, quinoline 6-C), 128.3 (quinazoline 8-C), 127.9 (quinazoline 6-C), 126.6 (quinazoline 5-C), 123.4 (d, JC–F = 4.8 Hz, quinoline 5-C), 122.4 (quinazoline 4a-C), 115.4 (d, JC–F = 18.3 Hz, quinoline 7-C), 40.5 (butyl 1-C), 31.5 (butyl 2-C), 20.4 (butyl 3-C), 13.9 (butyl 4-C). HRMS (ESI-TOF) m/z calcd. for C22H20FN4O2 ([M + H]+) 391.1565. Found 391.1561.
2-[3-(Morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (10). A mixture of Luotonin A (9) (0.029 g, 0.1 mmol), K2CO3 (0.014 g, 0.1 mmol) and morpholine (0.087 g, 1 mmol) in DMSO (5 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (20 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure to afford 10 (0.020 g, 52%) as almost colorless crystals, m.p. > 272 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 296 (21%), 211 (27), 111 (32), 109 (24), 97 (47), 95 (39), 85 (38), 83 (46), 81 (51), 71 (56), 69 (81), 67 (28), 57 (100); 1H-NMR (CDCl3) δ 11.15 (s, 1H, NH), 8.38 (dd, J = 7.9 Hz, 1.0 Hz, 1H, quinazoline 5-H), 8.25 (s, 1H, quinoline 4-H), 8.20 (d, J = 7.5 Hz, 1H, quinoline 8-H), 7.92 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.88 (ddd, J = 8.4 Hz, 6.9 Hz, 1.4 Hz, 1H, quinoline 7-H), 7.85–7.75 (m, 2H, quinazoline 7-H, 8-H), 7.72 (ddd, J = 8.2 Hz, 7.0 Hz, 1.2 Hz, 1H, quinoline 6-H), 7.57 (ddd, J = 8.2 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 6-H), 4.29 (dt, J = 13.4 Hz, 3.5 Hz, 1H, morpholine 3-H), 4.10 (dt, J = 11.6 Hz, 3.7 Hz, 1H, morpholine 2-H), 3.86–3.76 (m, 1H, morpholine 2′-H), 3.74–3.63 (m, 2H, morpholine 3′-H, 6-H), 3.55 (ddd, J = 11.7 Hz, 8.8 Hz, 3.2 Hz, 1H, morpholine 6′-H), 3.32–3.24 (m, 2H, morpholine 5-H, 5′-H); 13C-NMR (CDCl3) δ: 169.2 (amide C=O), 161.4 (quinazoline 4-C), 148.6 (quinazoline 8a-C), 148.1 (quinazoline 2-C), 146.5 (quinoline 8a-C), 144.3 (quinoline 2-C), 136.6 (quinoline 4-C), 134.9 (quinazoline 7-C), 131.7 (quinoline 7-C), 129.8 (quinoline 8-C), 129.5 (quinoline 6-C), 129.2 (quinoline 3-C), 128.5 (quinazoline 8-C), 128.4 (quinoline 4a-C), 128.2 (quinazoline 6-C), 127.9 (quinoline 5-C), 127.0 (quinazoline 5-C), 122.8 (quinazoline 4a-C), 66.6 (morpholine 2-C), 66.3 (morpholine 6-C), 47.3 (morpholine 5-C), 42.6 (morpholine 3-C), HRMS (ESI-TOF) m/z calcd. for C22H19N4O3 ([M + H]+) 387.1452. Found 387.1451.
N-Butyl-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (11). A mixture of Luotonin A (9) (0.057 g, 0.2 mmol), K2CO3 (0.028 g, 0.2 mmol) and n-butylamine (0.146 g, 2 mmol) in DMSO (7 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (30 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure to afford 11 (0.052 g, 70%) as orange crystals, m.p. > 227 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 373 ([M + 1]+, 2%), 372 (M+, 6), 329 (9), 301 (63), 300 (100), 273 (29), 153 (22), 119 (33), 90 (23); 1H-NMR (CDCl3) δ 11.00 (s, 1H, quinazolinone NH), 8.37 (s, 1H, quinoline 4-H), 8.27 (d, J = 8.2 Hz, 1H, quinazoline 5-H), 8.10 (d, J = 8.4 Hz, 1H, quinoline 8-H), 7.88 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.86–7.80 (m, 1H, quinoline 7-H), 7.79–7.71 (m, 2H, quinazoline 7-H, 8-H), 7.67 (t, J = 7.5 Hz, 1H, quinoline 6-H), 7.48 (ddd, J = 8.1 Hz, 6.2 Hz, 2.1 Hz, 1H, quinazoline 6-H), 6.50 (t, J = 5.2 Hz, 1H, amide NH), 3.69–3.57 (m, 2H, butyl 1-CH2), 1.80–1.68 (m, 2H, butyl 2-CH2), 1.49 (dq, J = 14.8 Hz, 7.4 Hz, 2H, butyl 3-CH2), 1.00 (t, J = 7.4 Hz, 3H, CH3); 13C-NMR (CDCl3) δ: 168.6 (amide C=O), 161.5 (quinazoline 4-C), 148.5 (quinazoline 8a-C), 148.1 (quinazoline 2-C), 146.4 (quinoline 8a-C), 144.5 (quinoline 2-C), 137.9 (quinoline 4-C), 134.7 (quinazoline 7-C), 131.6 (quinoline 7-C), 130.6 (quinoline 3-C), 129.6 (quinoline 8-C), 129.3 (quinoline 6-C), 128.4 (quinazoline 8-C), 128.1 (quinoline 4a-C), 128.0 (quinazoline 6-C), 127.9 (quinoline 5-C), 126.8 (quinazoline 5-C), 122.6 (quinazoline 4a-C), 40.6 (butyl 1-C), 31.6 (butyl 2-C), 20.5 (butyl 3-C), 14.0 (butyl 4-C). HRMS (ESI-TOF) m/z calcd. for C22H20N4NaO2 ([M + Na]+) 395.1478. Found 395.1476.
N-[2-(Dimethylamino)ethyl]-2-(4-oxo-3,4-dihydroquinazolin-2-yl)quinoline-3-carboxamide (12). A mixture of Luotonin A (9) (0.100 g, 0.35 mmol), K2CO3 (0.048 g, 0.35 mmol) and N,N-dimethylethylenediamine (0.309 g, 3.5 mmol) in DMSO (10 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, dried over Na2SO4 and evaporated under reduced pressure to afford 12 as crude product (0.070 g, 52%). The residue was subjected to MPLC, eluting with CH2Cl2/methanol (80 + 20) to afford 12 as colorless crystals, m.p. 185–188 °C (CH2Cl2). MS (EI, 70 eV) m/z = 197 (5%), 155 (18), 137 (46), 110 (34), 108 (12), 83 (14), 82 (13), 57 (13), 43 (100); 1H-NMR (CDCl3) δ 11.15 (br s, 1H, quinazolinone NH), 8.37 (s, 1H, quinoline 4-H), 8.35 (ddd, J = 8.0 Hz, 1.2 Hz, 0.6 Hz, 1H, quinazoline 5-H), 8.15 (d, J = 8.4 Hz, 1H, quinoline 8-H), 7.88 (d, J = 8.1 Hz, 1H, quinoline 5-H), 7.84 (ddd, J = 8.4 Hz, 6.9 Hz, 1.4 Hz, 1H, quinoline 7-H), 7.80–7.73 (m, 2H, quinazoline 7-H, 8-H), 7.67 (ddd, J = 8.1 Hz, 7.0 Hz, 1.1 Hz, 1H, quinoline 6-H), 7.52 (ddd, J = 8.2 Hz, 6.3 Hz, 2.0 Hz, 1H, quinazoline 6-H), 6.81 (br s, 1H, amide NH), 3.72 (q, J = 5.7 Hz, 2H, 1-CH2), 2.62 (t, J = 5.7 Hz, 2H, 2-CH2), 2.19 (s, 6H, CH3); 13C-NMR (CDCl3) δ: 168.8 (amide C=O), 161.5 (quinazoline 4-C), 148.6 (quinazoline 8a-C), 148.2 (quinazoline 2-C), 146.5 (quinoline 8a-C), 144.7 (quinoline 2-C), 137.7 (quinoline 4-C), 134.7 (quinazoline 7-C), 131.6 (quinoline 7-C), 130.5 (quinoline 3-C), 129.6 (quinoline 8-C), 129.2 (quinoline 6-C), 128.4 (quinazoline 8-C), 128.1 (quinaoline 4a-C), 128.0 (quinoline 5-C or quinazoline 6-C), 127.9 (quinazoline 6-C or quinoline 5-C), 126.9 (quinazoline 5-C), 122.7 (quinazoline 4a-C), 57.5 (2-CH2), 45.1 (CH3), 37.9 (1-C7H2). HRMS (ESI-TOF) m/z calcd. for C22H22N5O2 ([M + H]+): 388.1768. Found: 388.1772.
2-[5,7-Dimethoxy-3-(morpholin-4-ylcarbonyl)quinolin-2-yl]quinazolin-4(3H)-one (14). A mixture of 1,3-dimethoxyquinolino[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (13) (0.172 g, 0.5 mmol), K2CO3 (0.069 g, 0.5 mmol) and morpholine (0.436 g, 5 mmol) in DMSO (15 mL) was stirred at 100 °C for 16 h, then it was cooled to room temperature and the volatile components were removed by Kugelrohr distillation (10−1 mbar, 100 °C). The residue was taken up in CH2Cl2 (50 mL) and the solution was washed with water and brine, then dried over Na2SO4 and evaporated under reduced pressure. The residue was subjected to MPLC, eluting with CH2Cl2/methanol (95 + 5) to afford 14 (0.060 g, 27%) as yellowish crystals, m.p. > 264 °C (sublimation; CH2Cl2). MS (EI, 70 eV) m/z = 447 ([M + 1]+, 2%), 446 (M+, 8) 361 (78), 360 (100), 333 (42), 302 (22), 119 (26), 90 (32), 86 (25), 57 (30), 56 (57), 55 (27); 1H-NMR (CDCl3) δ 11.14 (s, 1H, NH), 8.50 (d, J = 0.5 Hz, 1H, quinoline 4-H), 8.37 (dd, J = 8.0 Hz, 1.1 Hz, 1H, quinazoline 5-H), 7.80 (ddd, J = 8.3 Hz, 6.8 Hz, 1.5 Hz, 1H, quinazoline 7-H), 7.76 (dd, J = 8.1 Hz, 1.0 Hz, 1H, quinazoline 8-H), 7.55 (ddd, J = 8.2 Hz, 6.9 Hz, 1.5 Hz, 1H, quinazoline 6-H), 7.06 (d, J = 1.6 Hz, 1H, quinoline 8-H), 6.63 (d, J = 2.1 Hz, 1H, quinoline 6-H), 4.29–4.24 (m, 1H, morpholine 3-H), 4.11–4.06 (m, 1H, morpholine 2-H), 4.02 (s, 3H, 7-OCH3), 4.01 (s, 3H, 5-OCH3), 3.84–3.77 (m, 1H, morpholine 2′-H), 3.73–3.66 (m, 2H, morpholine 3′-H, 6-H), 3.59–3.52 (m, 1H, morpholine 6′-H), 3.31–3.23 (m, 2H, morpholine 5-H, 5′-H); 13C-NMR (CDCl3) δ: 169.7 (amide C=O), 163.1 (quinoline 7-C), 161.4 (quinazoline 4-C), 156.1 (quinoline 5-C), 148.9 (quinoline 8a-C), 148.7 (quinazoline 8a-C), 148.3 (quinazoline 2-C), 144.6 (quinoline 2-C), 134.8 (quinazoline 7-C), 131.7 (quinoline 4-C), 128.5 (quinazoline 8-C), 128.0 (quinazoline 6-C), 127.0 (quinazoline 5-C), 126.0 (quinoline 3-C), 122.8 (quinazoline 4a-C), 117.4 (quinoline 4a-C), 100.7 (quinoline 6-C), 99.5 (quinoline 8-C), 66.7 (morpholine 2-C), 66.4 (morpholine 6-C), 56.2 (7-OCH3), 56.0 (5-OCH3), 47.3 (morpholine 5-C), 42.6 (morpholine 3-C). HRMS (ESI-TOF) m/z calcd. for C24H23N4O5 ([M + H]+) 447.1663. Found 447.1662.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}