3.1. By Metal-Catalyzed Cycloadditions
Acylhydrazones can be visualized as imine equivalents, and their benzoyl derivatives have been shown to react with several nucleophiles under Lewis acid catalytic conditions or in the presence of Lewis base promoter. Acylhydrazones have also been explored as general 1,3-dipoles, and it has been found that the [3+2] cycloaddition reaction of acylhydrazones with olefins is possible under Lewis acid catalytic conditions [
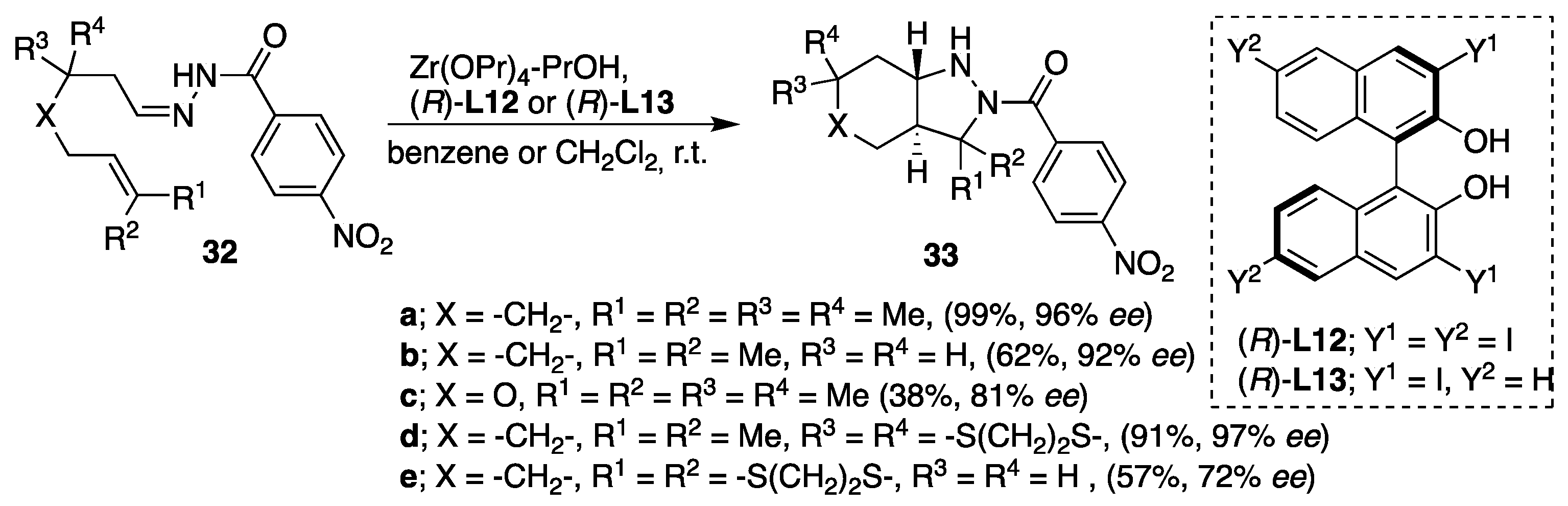
28]. Additionally, asymmetric intramolecular [3+2] cycloadditions of acylhydrazones have been described by Kobayashi using chiral zirconium catalyst leading to condensed pyrazolidine derivatives [
29]. The chiral zirconium catalyst was prepared in situ from Zr(OPr)
4 (10 mol %) and selected (
R)-BINOL ligand (12 mol %). The reaction was run in benzene or dichloromethane in the presence of 50 mol % of PrOH as an additive at room temperature yielding the corresponding fused pyrazolidine derivatives as major
trans isomers (
cis/
trans ratio > 1/99) and with
ee from 72 up to 97% (
Scheme 11). Although that precise reaction pathway in these reactions is not clarified, it was clearly shown that
cis/
trans selectivity was controlled by the chirality of the chiral catalyst. For example, when (
S)-
L12 was used as a catalyst instead of (
R)-
L12 under the same reaction conditions, the corresponding
cis isomer of cycloadduct of (
S)-citronellal-derived 4-nitrobenzoylhydrazone was obtained with good selectivity.
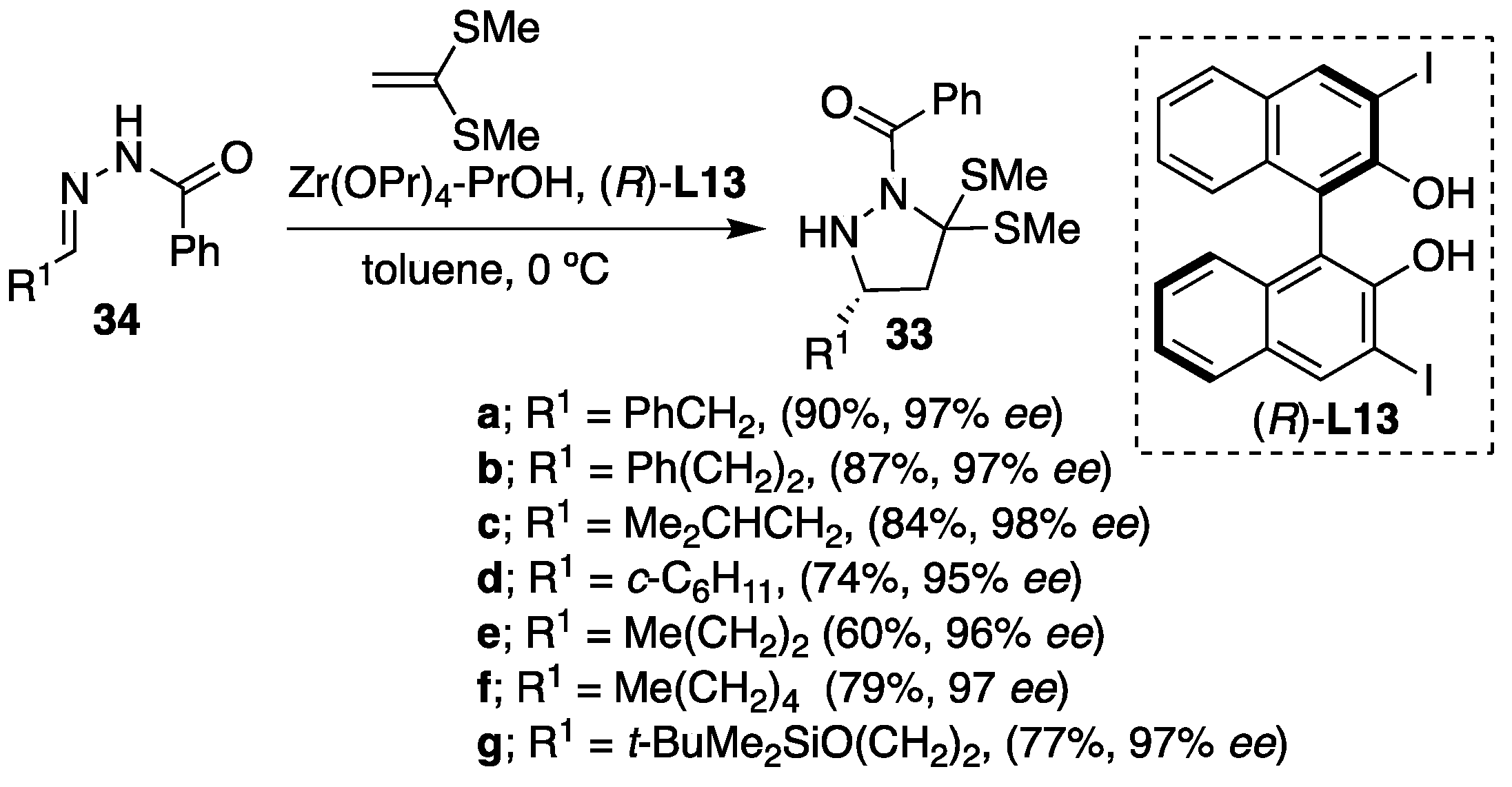
However, in this methodology, the substrate scope was restricted, and the reactions were limited to only an intramolecular type of [3+2] cyclisation. Later on, the same group developed the efficient zirconium-catalyzed enantioselective [3+2] intermolecular cycloaddition of hydrazones to olefins leading to optically-active pyrazolidine, pyrazoline and 1,3-diamine derivatives [
30]. In this case, benzoylhydrazones were found to be superior substrates over the 4-nitrobenzoyl analogues. The amount of olefin used also effected the reactivity, and the yields were significantly improved by using two equiv. of substituted olefins. Hydrazones derived from a β-branched aldehyde, a sterically-hindered aldehyde, enolizable aldehydes and functionalized aldehydes all readily reacted with ethene-1,1-diylbis(methylsulfane) without any side reactions, and the corresponding substituted pyrazolidines
35 were obtained in high yield and high levels of enantioselectivity (
Scheme 12).
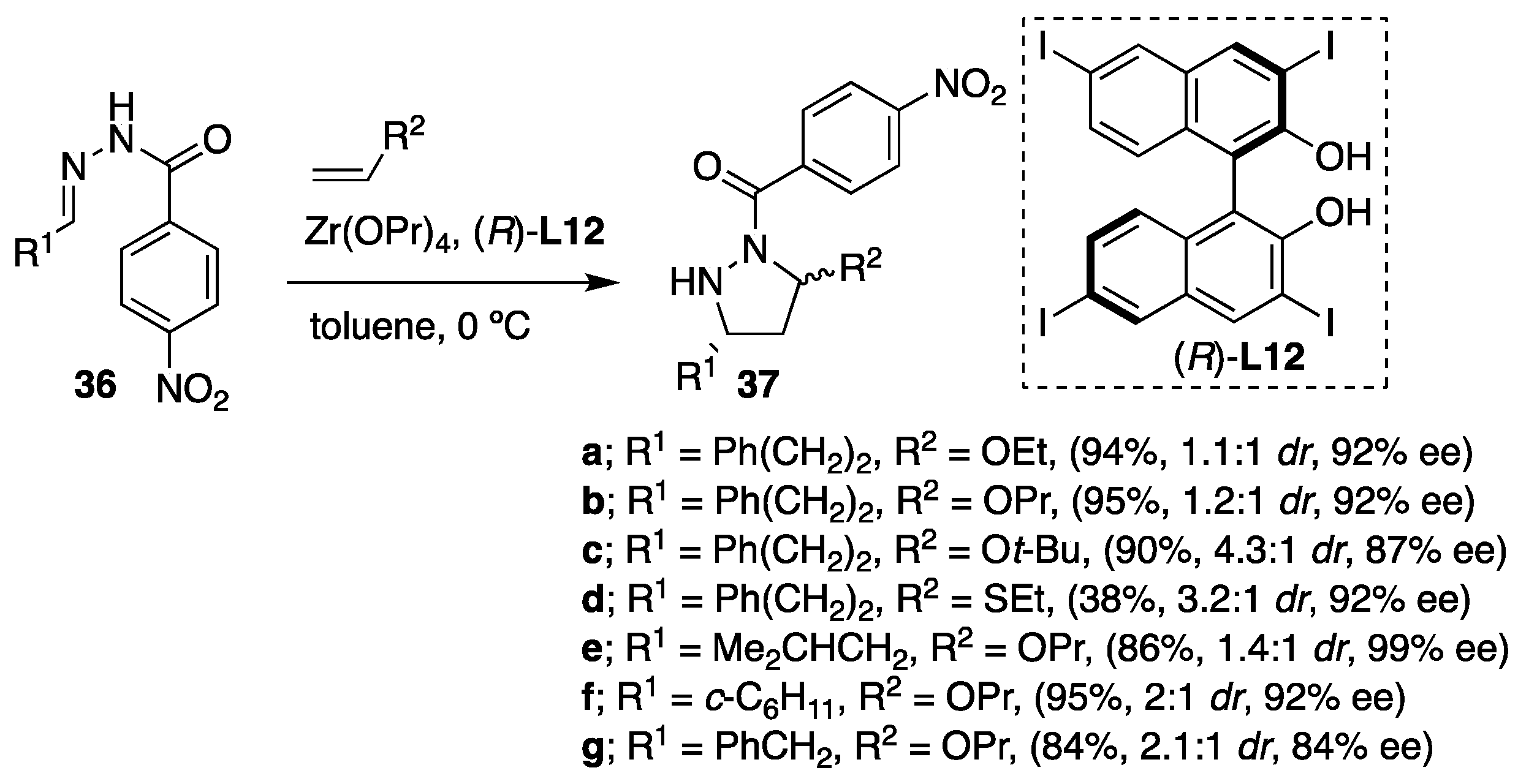
Furthermore, the variety of hydrazones derived from α-branched and β-branched aliphatic aldehydes reacted readily with different vinyl ethers as olefins furnishing the pyrazolidines
37 containing a
N,
O-acetal functionality in high yields and enantioselectivities (
Scheme 13). In this particular case,
p-nitrobenzoyl hydrazones were found to be more suitable derivatives than their benzoyl analogues due to their higher reactivity under the catalytic conditions. However, moderate diastereoselectivities were obtained in the majority of the cases (diastereomer ratio (
dr) > 1/1), whereas both diastereomers showed excellent enantioselectivity in most instances (
ee > 81%). Concerning the mechanistic aspect of the transformations, the authors suggested a concerted reaction pathway over the alternative stepwise pathway. The mechanism of the transformation was among the other facts elucidated on the observation that both diastereomers of the products have the same absolute configuration at the carbon atom bearing the R
1 substituent (
Scheme 13).
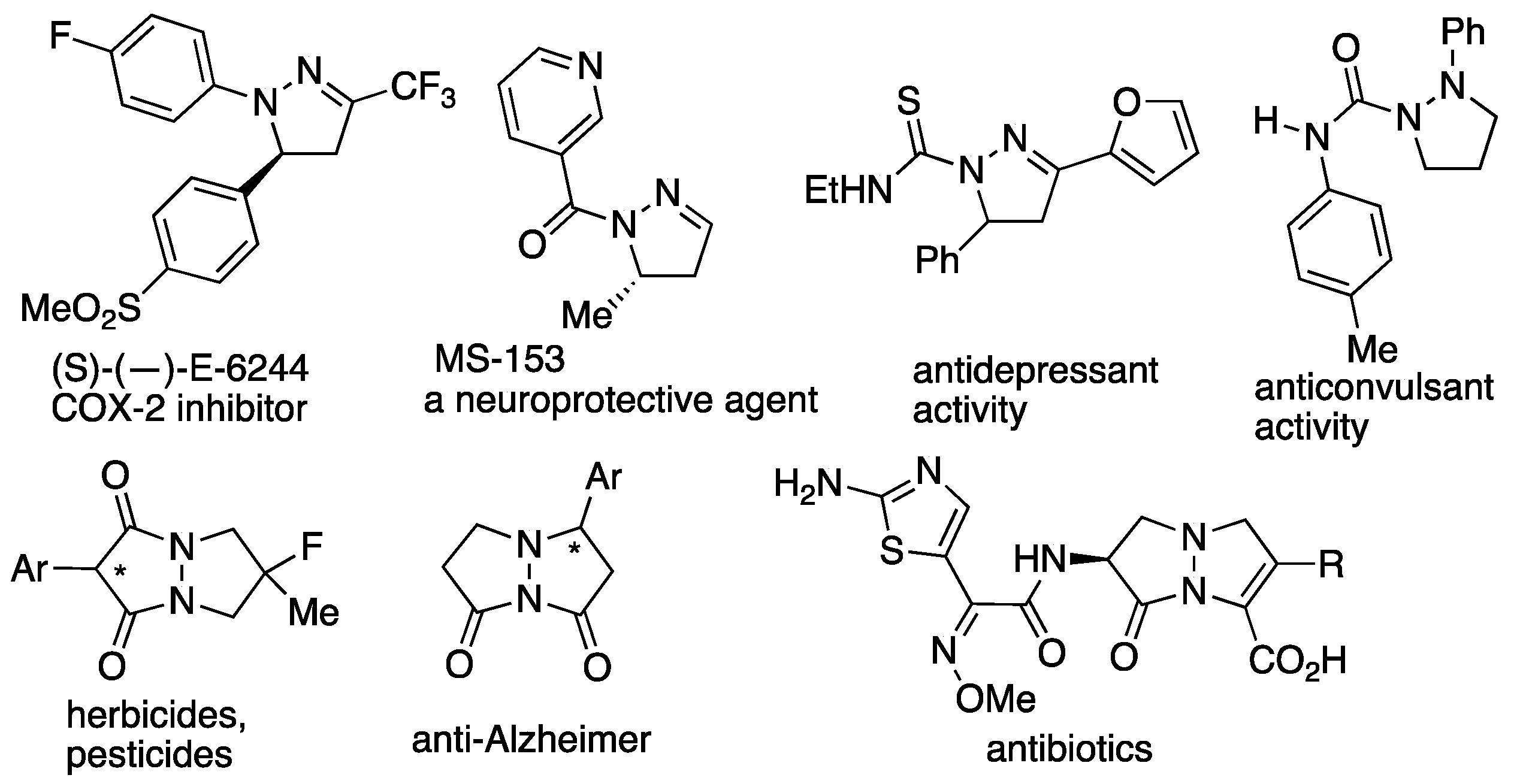
The developed methodology was elegantly applied for the synthesis of enantioenriched compound
MS-153 of biological importance. The [3+2] cycloaddition of hydrazone
38 to propyl vinyl ether proceeded with 76% yield and 84%
ee for the major and 97%
ee for the minor isomer. The
p-nitrobenzoyl functionality was removed by treatment with LiAlH
4 at −78 °C in THF followed by nicotinoylation. The obtained intermediate
40 was a useful starting material for the synthesis of
MS-153 derivatives. However, after the removal of the phenylthio group using Raney-Ni,
ent MS-153 was obtained with high enantioselectivity (88%
ee) (
Scheme 14).
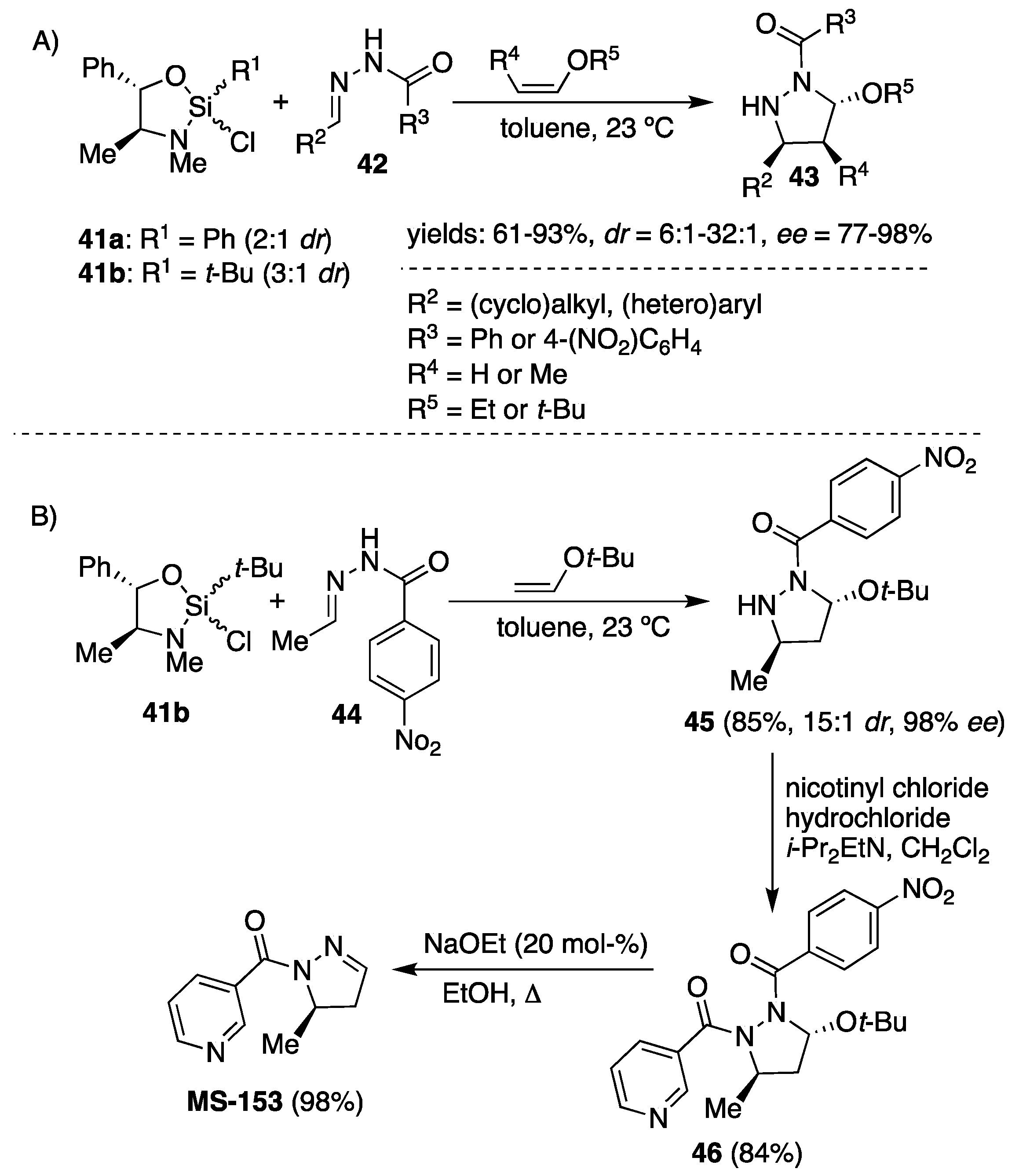
Phenyl and
tert-butylsilane reagents (easily prepared from pseudoephedrine and phenyl or
tert-butyltrichlorosilane as a mixture of diastereomers at silicon) introduced by Leigghton et al. were applied for acylhydrazone-enol ether cycloadditions [
31]. Phenylsilane was found to facilitate the cycloaddition of
N′-(3-phenylpropylidene)benzohydrazide with ethoxyethene to yield the corresponding pyrazolidine in 61% with 6:1 diastereoselectivity and 77%
ee. The use of
tert-butyl vinyl ether resulted in significant improvement in both diastereoselectivity (
dr 24:1) and enantioselectivity (90%
ee), and the transformation was found to perform the best in toluene at room temperature (
Scheme 15). Variety of benzoylhydrazones derived from aliphatic (Ph(CH
2)
2CHO, BnOCH
2CHO, CyCHO,
t-BuCHO) and (hetero)aromatic (benzaldehyde, 4-fluorobenzaldehide and furan-2-carbaldehyde) aldehydes reacted readily with
tert-butyl vinyl ether (three equiv.) in the presence of phenylsilane (1.5 equiv.), yielding 3,5-disubstituted pyrazolidines
43 up to 85% yield, >19:1
dr and
ee’s above 90% (
Scheme 15). Unlike the Zr(II)-catalyzed cycloaddition of benzoylhydrazones
36 (
Scheme 13), in this case, the β-substituted enol ethers reacted highly stereoselectively (>10:1
dr, 97%
ee) with
N′-(3-phenylpropylidene)benzohydrazides
42 providing enantio-enriched 3,4,5-trisubstituted pyrazolidine
43 (
Scheme 15, Example A). Selectivity with the acetaldehyde-derived hydrazone (R
2 = Me,
Scheme 15) is significantly reduced relative to that observed with larger R
2 groups when phenylsilane
41a is applied. Replacing the phenyl moiety in chiral silicon Lewis acid
41a with sterically more demanding
tert-butyl group
41b resulted in significant improvement in stereoselectivity when more electrophilic
N′-ethylidene-4-nitrobenzohydrazide
44 was reacted with
tert-butyl vinyl ether to provide (5
R)-3-(
tert-butoxy)-5-methylpyrazolidine
45 (
Scheme 15, Example B) in good yield of 85% and stereoselectivity (>15:1
dr, 98%
ee). Two additional synthetic steps furnished the targeted
MS-153 substrate in 70% overall yield and > 99%
ee (
Scheme 15, Example B) [
32]. Furthermore, a concise synthesis of manzacidin C based on the second generation [
33] chiral silicon Lewis acid, which promoted diastereo- and enantioselective acylhydrazone-alkene [3+2] cycloaddition, has also been reported by the same group [
34]. The corresponding enantioenriched 3,5,5-trisubstituded pyrazolidine precursor was subsequently used for the synthesis of crucial 1,3-bisamide intermediate, which was obtained in 73% yield and high stereoselectivity (>20:1
dr and 94%
ee).
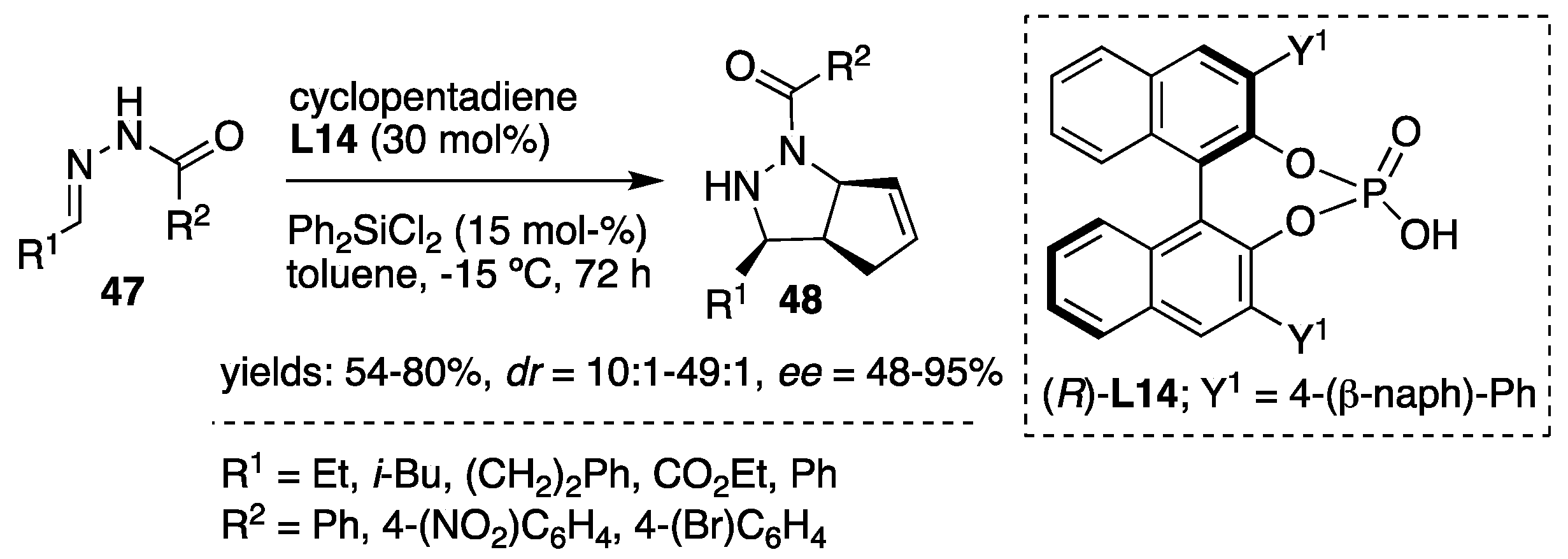
In addition to the above-described silicon Lewis acid-promoted [3+2] cycloadditions of acylhydrazones, Tsogoeva et al. [
35] developed a catalytic system comprised of chiral Brønsted acid (chiral BINOL phosphates) and silicon Lewis acid (R
2SiX
2). The combination of both acids acted cooperatively in the stereoselective intermolecular cycloaddition of acylhydrazones with cyclopentadiene providing a convenient and facile process for the synthesis of pyrazolidines. Assumed synergism between the BINOL phosphate (
R)-
L14 and Ph
2SiCl
2 was also tested. Lewis acid (dichlorodiphenylsilane) alone was inactive in catalysis, whereas BINOL phosphate itself showed low reactivity and moderate enantioselectivity (99:1
dr, 47%
ee, example: R
1 = Et, R
2 = 4-(NO
2)-C
6H
4,
Scheme 16). The combination of both components in ratio (
R)-
L14/ Ph
2SiCl
2 in 2/1 was established to be the most active, producing the corresponding pyrazolidines
48 as major
cis isomers in good to excellent enantioselectivities (
Scheme 16).
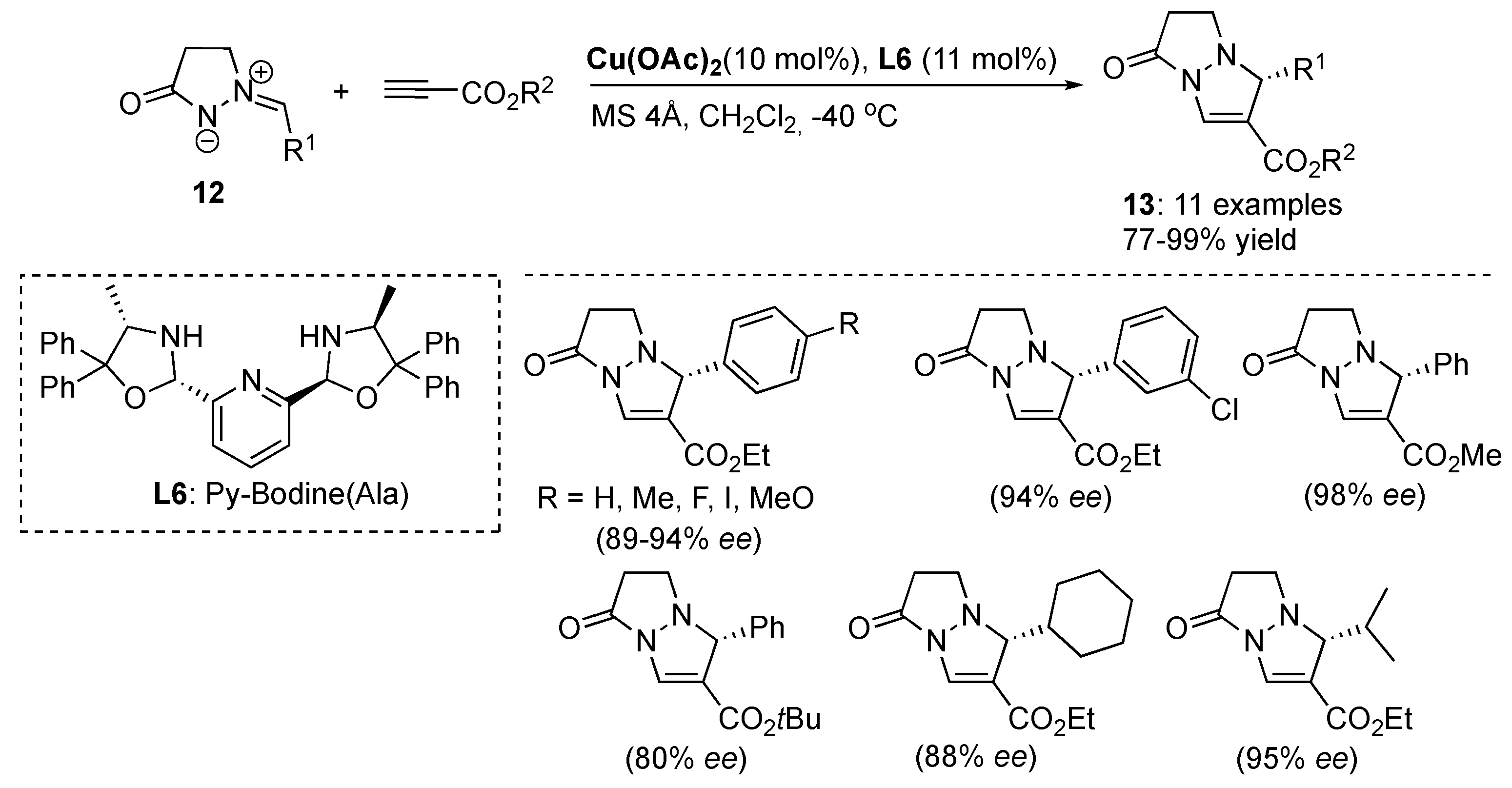
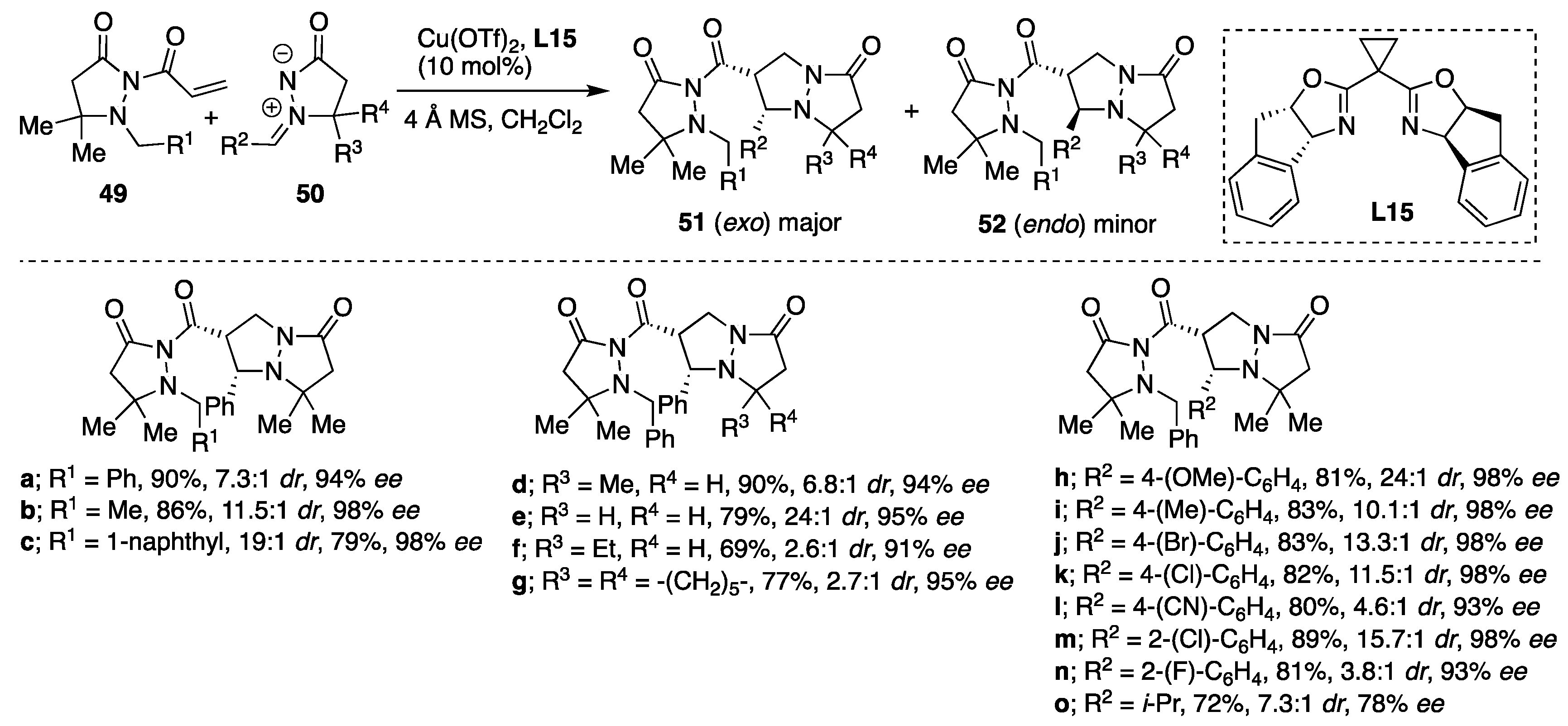
Copper(II)-catalyzed diastereo- and enantioselective cycloadditions of azomethine imines to 2-acryloyl-3-pyrazolidinones were developed by Sibi et al. deriving the corresponding
exo cycloadducts with high diastereoselectivities (up to > 96:4
cis/
trans) and enantioselectivities for
cis isomer up to 98%
ee [
36]. The Cu(OTf)
2/
L15-catalytic system was found to be the most efficient in producing the corresponding cycloadducts
51 as a major
syn diastereomers and with excellent enantioselectivities (
Scheme 17). Chiral Lewis acid prepared from Cu(OTf)
2 and additional bis(oxazoline) ligands such as
t-Bu-BOX, Ph-BOX and Bn-BOX were also studied as catalysts; however, when the Cu(OTF)
2/
t-Bu-BOX system was used as a catalyst,
syn products were formed in good yields and with lower diastereoselectivity. On the other hand, Cu(OTF)
2/Ph-BOX and Cu(OTF)
2/Bn-BOX catalytic systems behaved similarly yielding
exo/
endo product in ~5/1 and 82%
ee of the major isomer. When applying the Cu(OTf)
2/
L15-catalytic system, generally excellent yields and good diastereo- and high enantioselectivities were possible from cycloadditions of a variety of C5 and
N′-arylmethylidene-substituted azomethine imines
50 and different α,β-unsaturated pyrazolidinone imides
49 (
Scheme 17). However, when the methodology was extended to β-substituted α,β-unsaturated pyrazolidinone imides such as pyrazolidinone chrotonate, no reaction was observed or the cycloadducts were formed in moderate yields and in low enantioselectivities.
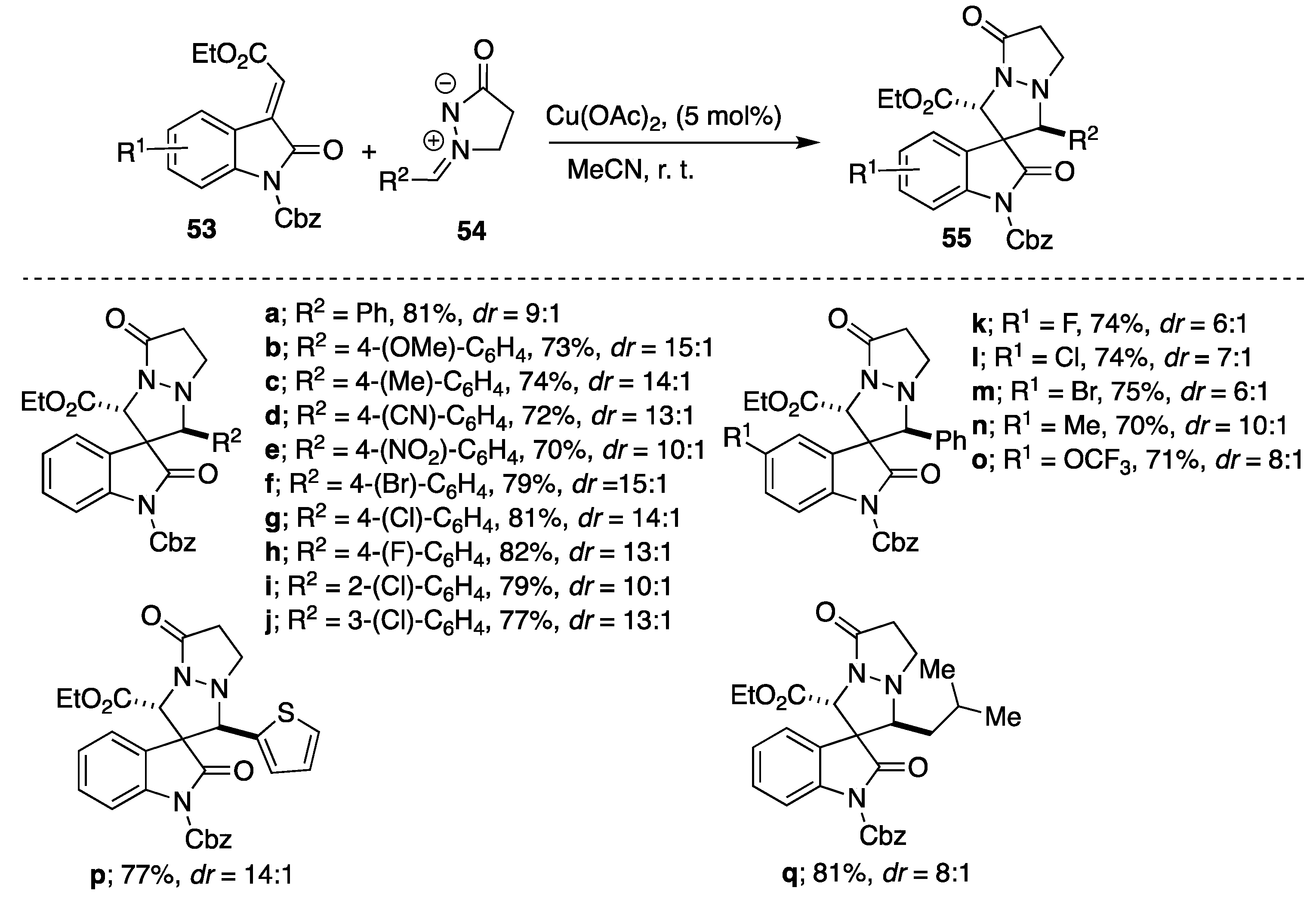
An additional example of copper(II)-catalyzed [3+2] cycloadditions of
N,
N′-cyclic azomethine imines is Cu(OAc)
2-catalyzed cycloaddition of azomethine imines to methyleneindolinones described by Sun and coworkers [
37]. Among tested Lewis acids (Cu(OAc)
2, AgOTf, Sc(OTf)
3, Zn(OTf)
2, CuI, Cu(acac)
2 and Cu[MeCN]
4PF
6), Cu(OAc)
2 was found to be the most efficient in acetonitrile as a solvent of choice. Generally, when Cu(OAc)
2 was used as a Lewis acid catalyst, the reactions proceeded smoothly to afford the desired spiro adducts
55 in moderate to high yields (70–82%) and good stereoselectivities (9:1 up to 15:1) forming
trans diastereomer as the major isomer (
Scheme 18). Notably, the heteroaryl (2-thienyl) and alkyl substituted azomethine imines were tolerated in the reaction and derived the corresponding products
55p and
55q in 77%, 14:1
dr and 81%, 8:1
dr, respectively (
Scheme 18). An enantioselective variant of this reactions was also carried out screening a series of chiral ligands (
i-Pr- and
t-Bu-substituted oxazoline ligands and
i-Pr-Phosferrox ligand), affording the corresponding product
55a in moderate yield and low enantioselectivity (up to 48%
ee).
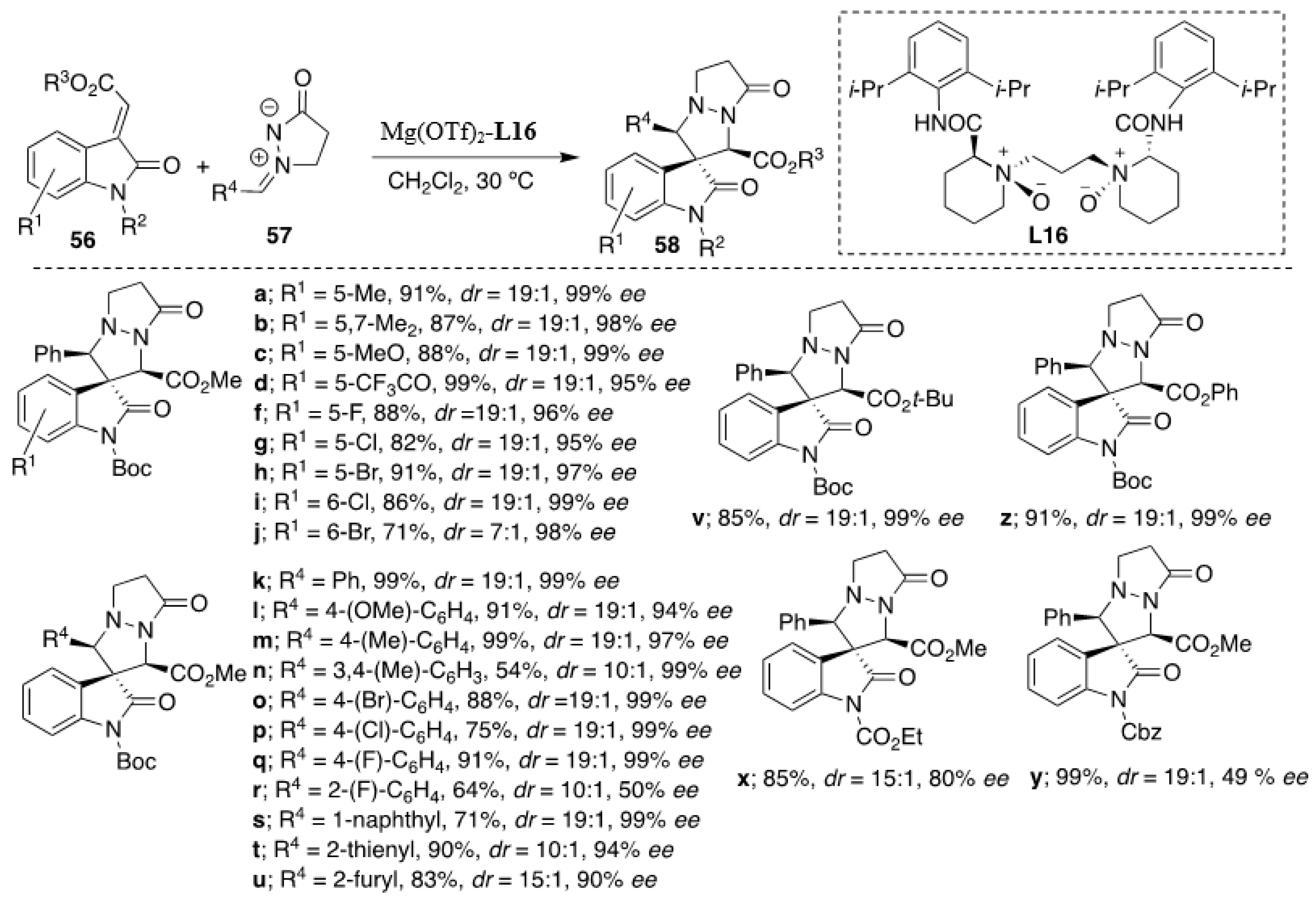
Recently, Feng et al. developed highly diastereo- and enantioselective [3+2] cycloaddition of methyleneindolinones with
N,
N′-cyclic azomethine imines, which was successfully catalyzed by a
N,
N′-dioxide-ligated-Mg(OTf)
2 complex [
38]. It should be stressed that unlike in the above-described Cu(OAc)
2-catalyzed examples [
37], in this case, the major
cis diastereomer prevailed. Three different alkaline metal salts (Ca(OTf)
2, Ba(ClO
4)
2, and Mg(OTf)
2 were tested in combination with several chiral
N,
N′-dioxide ligands derived from (
S)-proline, (
S)-Ramipril and (
S)-pipecolic acid. However, the complex of Mg(OTf)
2 could offer only one isomer in nearly quantitative yield in the test reaction. The optimized reaction conditions constituting of 5 mol % of catalyst (
L16-Mg(OTf)
2, 1:1) in dichloromethane at 30 °C in 48 h yielded the corresponding spiro adducts in excellent yields and enantioselectivities (
Scheme 19). Methyleneindolinones bearing substituents at different positions of the phenyl ring had little influence on the selectivity (
dr 19:1 in most cases with
ee’s 90–99%). Furthermore, good results were also obtained when changing the electronic and steric nature at
meta- or
para-position on the aryl moiety of the
N,
N′-cyclic azomethine imines. However, the azomethine imines with a substituent at the
ortho position in the phenyl ring in general gave lower yields of cycloadducts with decreased enantioselectivity (e.g., R
2 = 2-F-C
6H
4, 64% yield, 50%
ee,
Scheme 19). The relationship between the enantiomeric excess of the cycloadduct and
L16 showed a weak negative nonlinear effect, suggesting that both monomeric and oligomeric catalytic species exist in the solution, but the monomeric complex might act as the more active one since its existence was clearly confirmed by observing the molecular mass of [
L16 + Mg
2+ + OTf
−] in ESI-MS spectra.
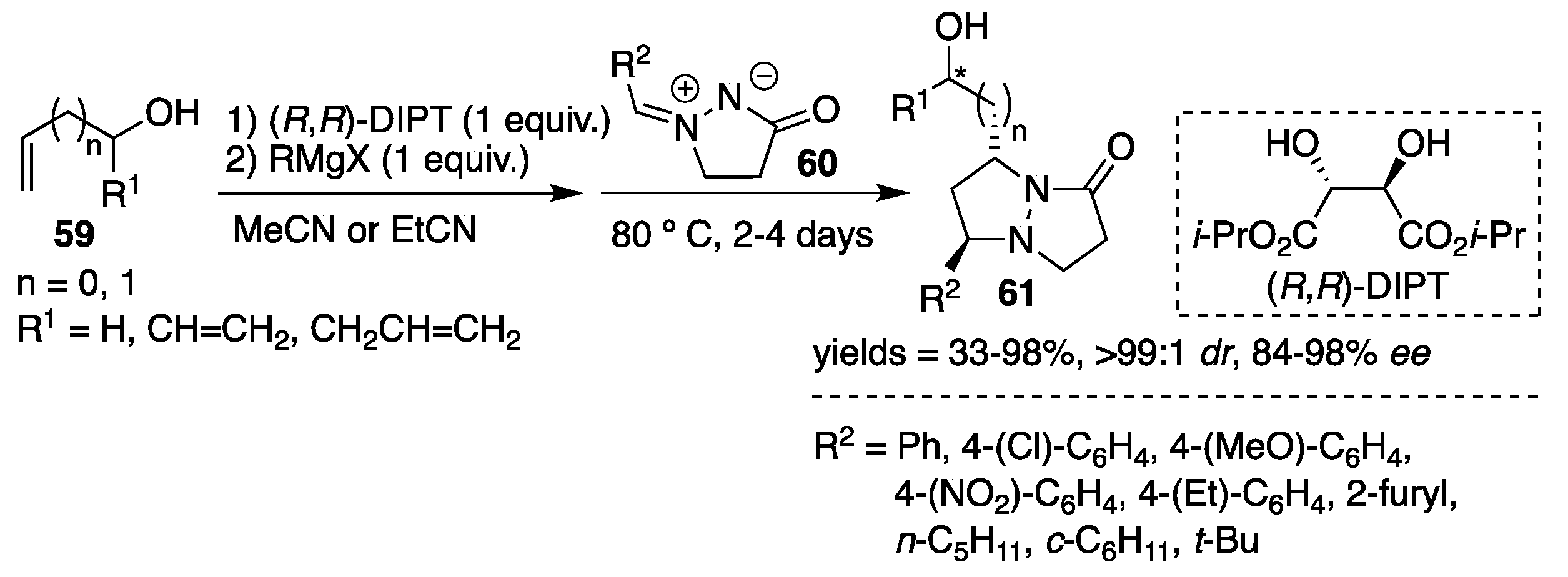
An attractive and unique 1,3-dipolar cycloaddition reaction of azomethine imines
60 to allylic [
39,
40] and homoallylic [
41] alcohols
59, based on a magnesium-mediated multinucleating chiral reaction system utilizing diisopropyl (
R,
R)-tartrate [(
R,
R)-DIPT] as a chiral reagent was developed by Ukaji and Inomata et al. The transformation was shown to be applicable to both aryl- and alkyl-substituted azomethine imines. The corresponding optically-enriched
trans-pyrazolidines
61 were obtained with excellent regio-, diastereo- and enantioselectivity, with results up to 98%
ee (
Scheme 20). As an example, a mixture of allyl alcohol and (
R,
R)-DIPT was reacted with alkylmagnesium halide as a magnesium source followed by the addition of azomethine imine. The reactions were performed at 80 °C and alkanonitriles (MeCN or EtCN) were found to be the most suitable solvents. The use of a catalytic amount of (
R,
R)-DIPT was also effective when accompanied by the addition of one equiv. of MgBr
2. The proposed transition state model consists of a carbonyl oxygen of azomethine imine, rather than imine nitrogen, coordinated to the magnesium salt of (
R,
R)-DIPT. In this model, the
pro-
R allyloxy moiety approaches the azomethine imine group, giving the
R,
R,
R-configuration (confirmed by the X-ray analysis of cycloadduct derivatives) of the cycloadducts.
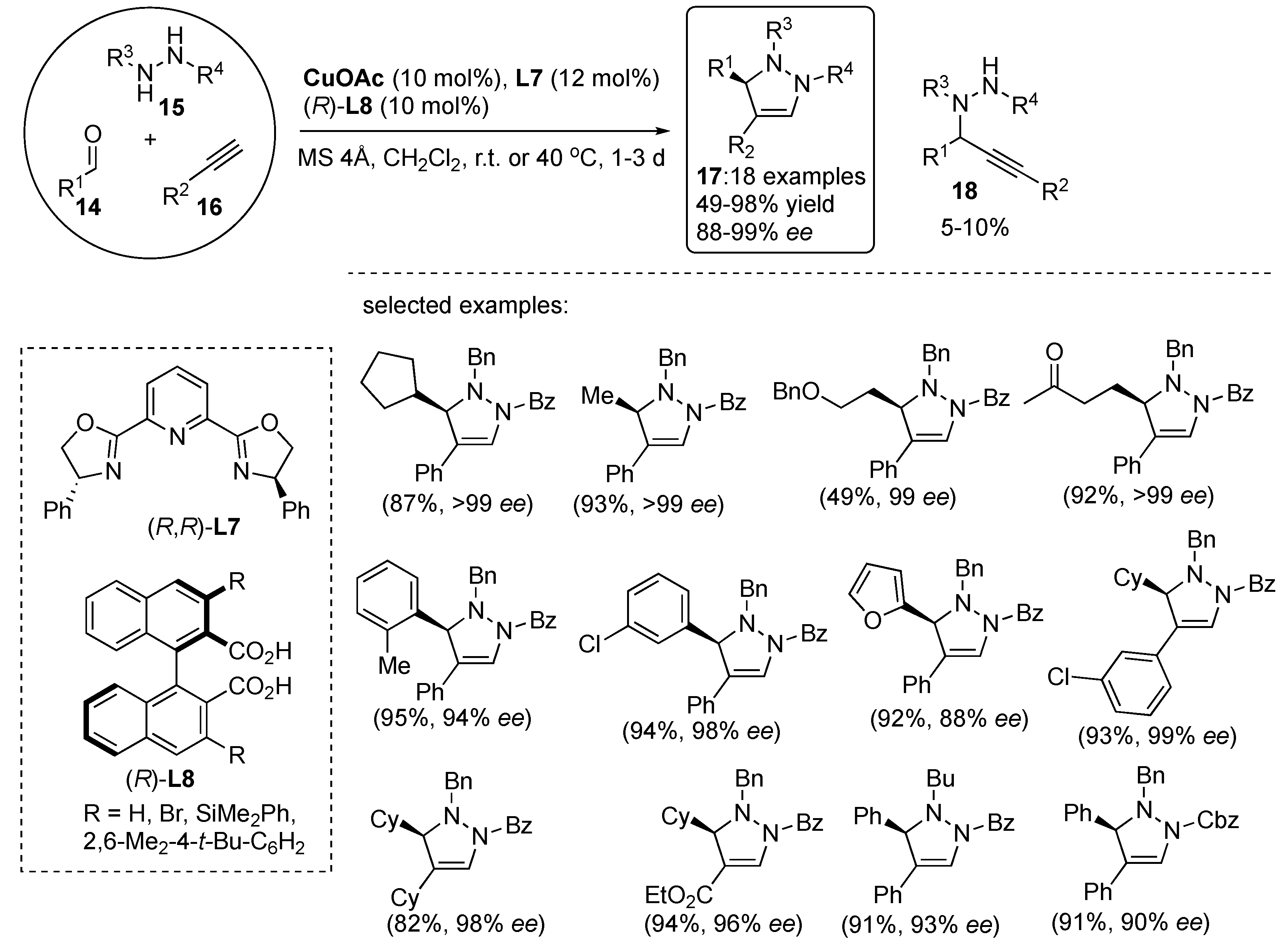
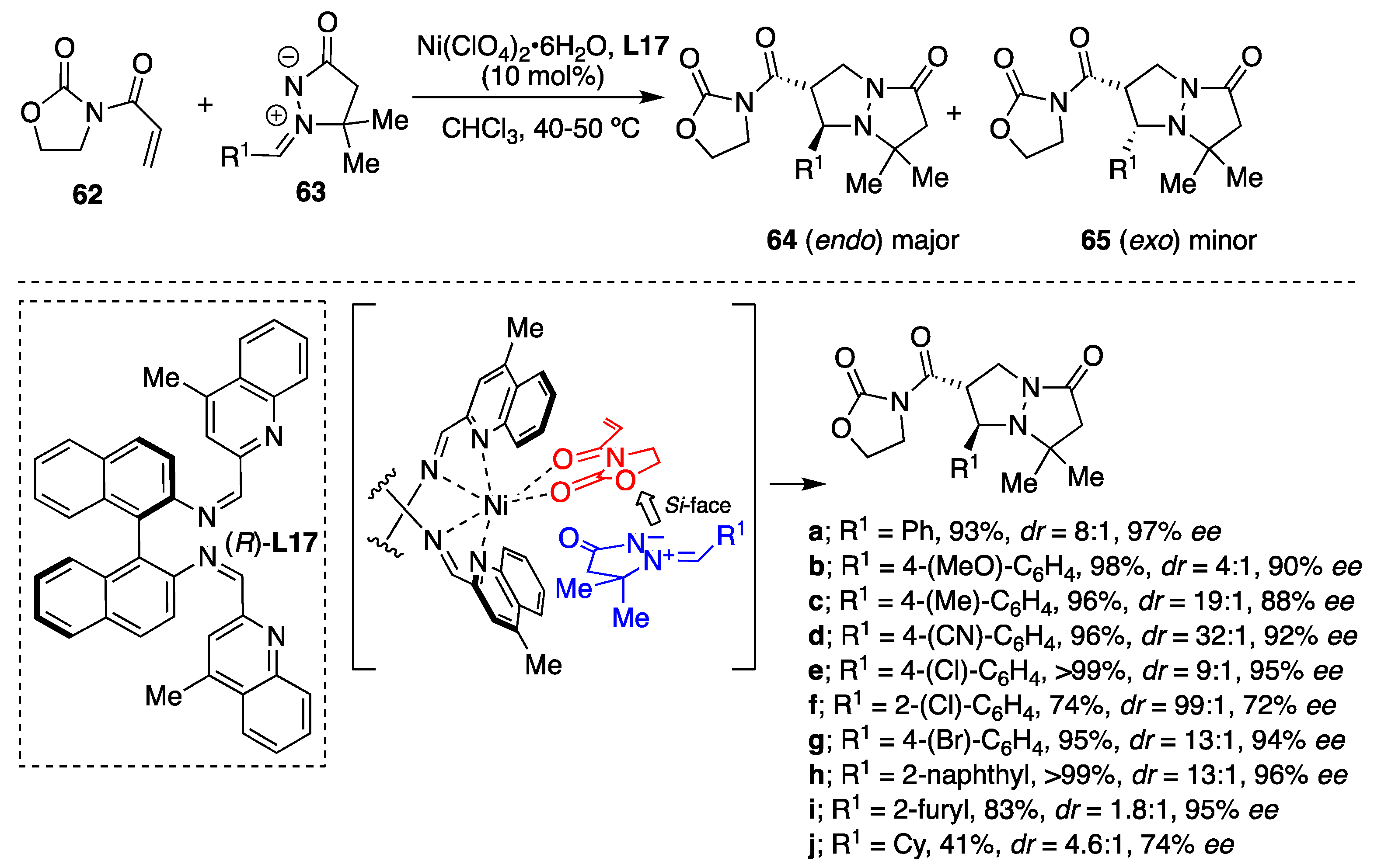
The diastereodivergent process leading to enantiopure pyrazolidine derivatives
64 and
65 from 3-acryloyl-2-oxazolidinone
62 and
N,
N′-cyclic azomethine imines
63 employing Ni(II)-catalyzed asymmetric [3+2] cycloadditions was developed [
42]. Among the tested catalytic systems, the in situ formed binaphthyl-quinolinediimine-based Ni(II) complex, (
R)-
L17–Ni(II), exhibited favorable activity yielding the corresponding pyrazolidine derivatives
64 as a major
trans diastereomer and with high enantioselectivity. Under the catalysis of (
R)-
L17–Ni(ClO
4)
2·6H
2O in chloroform at 40–50 °C and in the presence of molecular sieves (MS) 4 Å, the reaction proceeded well and was independent of the electronic character of the benzene ring substituents (
Scheme 21). It is of note here that the enantio- and diastereoselectivity improved in some cases upon increasing the reaction temperature from room temperature up to 50 °C, which could be attributed to enhanced ligand exchange rate of the transition state Ni(II)-complex. Although the reaction of C-cyclohexyl substituted azomethine imine resulted in moderate yield and 74%
ee, the cycloaddition of naphthyl and heteroaryl analogues afforded excellent enantioselectivities (
Scheme 21).
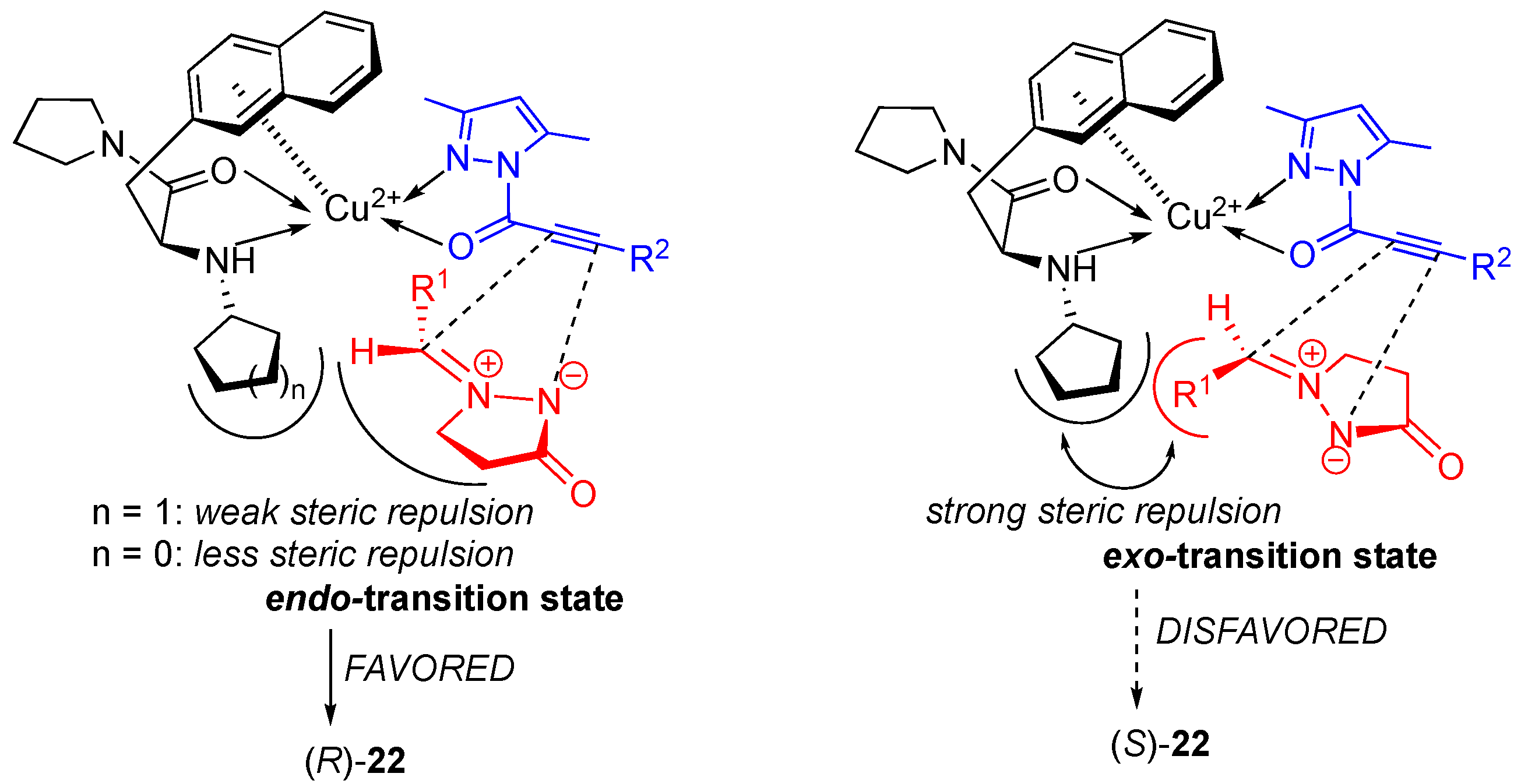
The results of the Ni(II)-catalyzed reaction are comparable to those of analogous Cu(II)-catalyzed reaction [
36], where
exo-cycloadduct (yielding the
cis-diastereomer) was the major product, complementarily since in these cases, the
endo-cycloaddition occurred. The difference in diastereoselectivity between the Cu(II)- and the Ni(II)-catalyzed cycloaddition of structurally-similar substrates can be explained by the alternative approach of the azomethine imine dipole depending on the metal geometry of catalytic chiral Lewis acid complex. The Cu(II)-catalyzed cycloaddition most probably proceeds via a distorted square-planar complex, which favors the
exo-approach of a dipole. However, the Ni(II)-catalyzed [3+2] cycloaddition likely proceeds through an octahedral complex forcing the
endo-approach of a dipole.
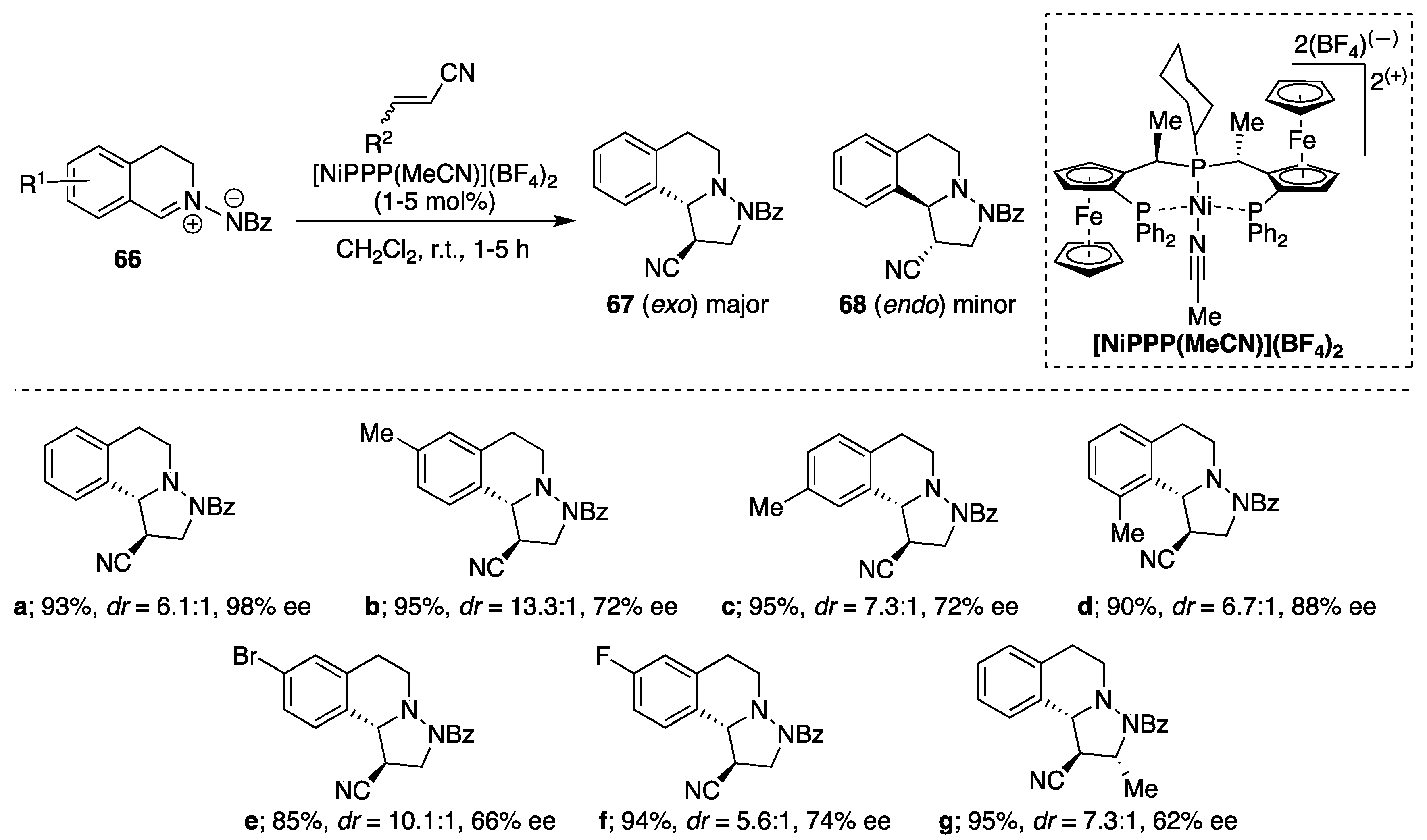
Togni et al. developed Ni(II)-Pigiphos enantioselective 1,3-dipolar cycloaddition of various
C,
N-cyclic azomethine imines
66 to α,β-unsaturated nitriles [
43]. When
N-benzoylamino-3,4-dihydroisoquinolinium betaine was reacted with acrylonitrile in the presence of 5 mol % of [Ni(PPP)(MeCN)](BF
4)
2 in dichloromethane, the completion of the reaction was observed after 0.5 h yielding 3,4-regioisomer, with not only good diastereomeric access of the
trans-isomer, but also excellent enantioselectivity (96%
ee). Under the optimized reaction conditions, the substrates functionalized with different electron-donating or electron-withdrawing groups yielded moderate enantiomeric excesses, form 60% up to 88%
ee (
Scheme 22). The
exo-selectivities were good to moderate, decreasing from 13.3:1 to 5.6:1 in decreasing the electron-donating ability of the substituent (Me, H, Br, F) on the position para to the 1,3-dipole. The relative
trans-stereoselectivity of the major cycloadduct
67 was established in each case by
1H and 2D NMR spectroscopy. Furthermore, the absolute configurations (3
R,4
R) could be established by X-ray crystallography of enantiopure single crystals of the major diastereomer. The potential influence of a more functionalized cyanoolefins on the
exo-selectivity of the transformation was also investigated using crotononitrile, methacrylonitrile,
trans-cinnamonitrile and
cis-2-pentenonitrile. The reactivity of this substituted acrylonitriles was much more inferior compared to the acrylonitrile, as no cycloaddition occurred with
trans-cinnamonitrile and
cis-2-pentenenitrile under the same reaction conditions. Furthermore, methacrylonitrile yielded 52% of the corresponding regioisomeric mixture of cycloadducts (3,4-cycloadduct/3,5-cycloadduct, 3.7/1) after 48 h at 40 °C. However, addition of crotononitrile (four equiv.,
cis/
trans mixture) to
N-benzoylamino-3,4-dihydroisoquinolinium betaine at r.t. over two days resulted largely in the formation of the
trans-3,4-cycloadduct (
dr = 7.3/1) in 66% yield and 62%
ee of the
trans-isomer (
Scheme 22, e.g.,
67g). On the basis of the above-mentioned results, it can be concluded that the steric size of the dipolarophile is a crucial factor in these 1,3-dipolar cycloadditions.
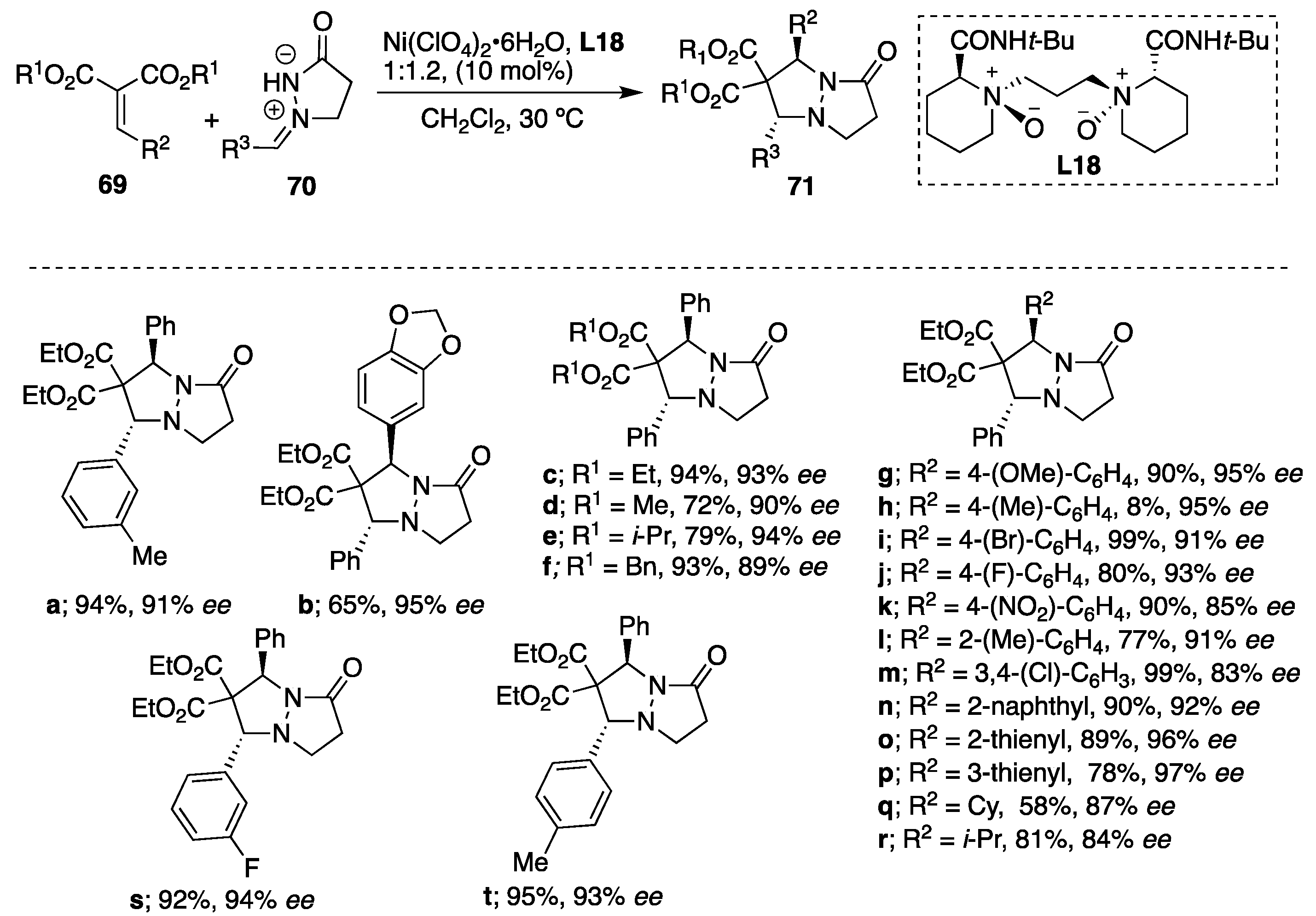
Feng et al. represented the first example of asymmetric reaction of azomethine imines
70 with alkylidene malonates
69 using
N,
N′-dioxide-Ni(II) complexes [
44]. Different metal sources such as Sc(OTf)
3, Mg(ClO
4)
2 and Co(ClO
4)
2·6H
2O were evaluated; however, the
N,
N′-dioxide-Ni(ClO
4)
2·6H
2O promoted the reaction giving rise to the corresponding endo-cycloadduct
71 as a single isomer (
dr > 99:1). Different nickel sources were also examined (Ni(BF
4)
2·6H
2O and NiBr
2), but no improvement in the cycloaddition was observed. The counter anion has a crucial effect on both the yield and the selectivity in the azomethine imine-alkylidene malonate cycloaddition. Upon the optimization of the reaction conditions, the ratio of 1:1.2 between Ni(ClO
4)
2·6H
2O and
N,
N′-dioxide ligand
L18 was chosen as optimal, and the reaction performed the best in dichloromethane. It is important to note that the solvent effects the reaction greatly since very low conversions were obtained in solvents such as toluene, THF, MeCN and EtOAc. The catalytic composition was also investigated, and a slightly positive nonlinear effect was obtained between the enantiomeric excess of the ligand and the product, suggesting that the minor oligomeric aggregation of the
N,
N′-dioxide-Ni(ClO
4)
2·6H
2O might exist in the catalytic system. However, the ESI-MS studies of Ni(ClO
4)
2·6H
2O and chiral ligand in a ratio of 1:1.2 in dichloromethane confirmed the existence of the monomeric nickel species [
L18 + Ni
(2+) + ClO
4−] in solution. Using the optimized catalytic system, a variety of dipolarophiles, (cyclo)alkyl- and (hetero)aryl-substituted alkylidene malonates, reacted with
N,
N′-cyclic azomethine imines, deriving the corresponding products as sole
trans-diastereomers with excellent enantioselectivities (from 83% up to 97%
ee) (
Scheme 23). To demonstrate the scalability, a gram-scale reaction was performed between diethyl 2-benzylidenemalonate and (
Z)-1-benzylidene-1λ
4-pyrazolidin-3-one under the optimized catalytic system to give the corresponding cycloadduct in 85% yield and 92%
ee.
3.2. By Organocatalyzed Cycloadditions
Rueping et al. efficiently employed acidic
N-triflylphosphoramide Brønsted acid
C1 as an organocatalyst in highly enantioselective cycloadditions between various alkenes and azomethine imines derived from
N-benzoylhydrazone precursors. Thus, under optimized reaction conditions, [3+2] cycloaddition of
N-benzoyl protected hydrazones
72 (derived from aliphatic, aromatic and heteroaromatic aldehydes or ethyl glyoxylate) to cyclopentadiene (
73) afforded pyrazolidine derivatives
74 in 51–99% yields with excellent stereoselectivity (
dr ≥ 96:4, 87–98%
ee). Next,
α-methylstyrene
75 and its (hetero)aryl analogues were used as dipolarophiles in [3+2] cycloaddition with
N-benzoyl-protected hydrazones
72, as azomethine imine precursors, furnishing valuable pyrazolidine derivatives
76, bearing a quaternary and a trisubstituted stereocenter at the 3- and 5-positions, in 52–95% yields and 80–96%
ee as single diastereomers (
Scheme 24) [
45].
Suga and co-workers reported cycloadditions of
N,
N-cyclic azomethine imines
77 to acrolein (
78) catalyzed by L-proline
C2 and (
S)-indoline-2-carboxylic acid
C3. The initial cycloadducts
8 were, after reduction with NaBH
4, isolated as the corresponding alcohols. Under the optimized reaction conditions, the L-proline
C2-catalyzed reactions gave the cycloadducts
79 in 54–89% yields as the major
endo-diastereomers (
endo/
exo = 83:27 to 99:1) with modest to good enantioselectivity (31–83%
ee). Azomethine imines bearing
o-chlorophenyl- and alkyl-substituents gave the corresponding products
79 in merely low to modest enantioselectivity (31–50%
ee). Interestingly, reactions catalyzed by (
S)-indoline-2-carboxylic acid
C3 furnished the corresponding cycloadducts
79 with high
exo-selectivity (
exo/
endo = 91:9 to 99:1) and good to excellent enantioselectivity (75–98%
ee), in 29–95% yields. Different substituents on the azomethine imines were well tolerated. The authors have shown that
exo-cycloadduct can be isomerized into
endo-cycloadduct in the presence of L-proline
C2 (
Scheme 25) [
46].
α,α-Bis[3,5-di(trifluoromethyl)phenyl]prolinol
C4 was applied as the organocatalyst of choice for the stereoselective cycloaddition of
N,
N-cyclic azomethine imines
80 to α,β-unsaturated aldehydes
81, furnishing the expected cycloadducts
82 in 50–95% yields with excellent stereoselectivities (
exo/
endo = 81:19 to 98:2; 82–97%
ee). Both electron-withdrawing and electron-donating substituents in the
para-position of benzene ring of azomethine imines gave the corresponding products with excellent stereoselectivities. Regarding the applied enals
81, the ones with an alkyl group worked very well, whereas reactions with cinnamaldehyde failed to give the expected products. Azomethine imine, derived from isobutyraldehyde, was successfully reacted with crotonaldehyde in the presence of MacMillan’s first generation organocatalyst
C5 [
47], giving the corresponding cycloadduct in 40% yield, 77%
ee and
exo/
endo = 95:5 (
Scheme 26) [
48].
Chen et al. applied 9-amino-9-deoxyepiquinine
C6 in the presence of 2,4,6-triisopropylbenzenesulfonic acid (TIPBA) for an efficient cycloaddition of various alkyl and (hetero)aryl substituted
N,
N-cyclic azomethine imines
84 to 2-cyclopenten-, 2-cyclohexen- and 2-cyclohepten-1-ones
83, giving the desired products
85 in excellent diastereoselectivity (
dr > 99:1), excellent enantioselectivity (85–95%
ee) and in 67–99% yields. When 9-amino-9-deoxyepiquinidine
C7 was applied, enantiomeric cycloadducts were obtained, retaining the same level of stereoselectivity (
Scheme 27). The 6-hydroxy group on the quinolone part of the organocatalysts
C6 and
C7, as well as the presence of the sterically bulky TIPBA additive were essential for the high stereoselectivity of these transformations [
49].
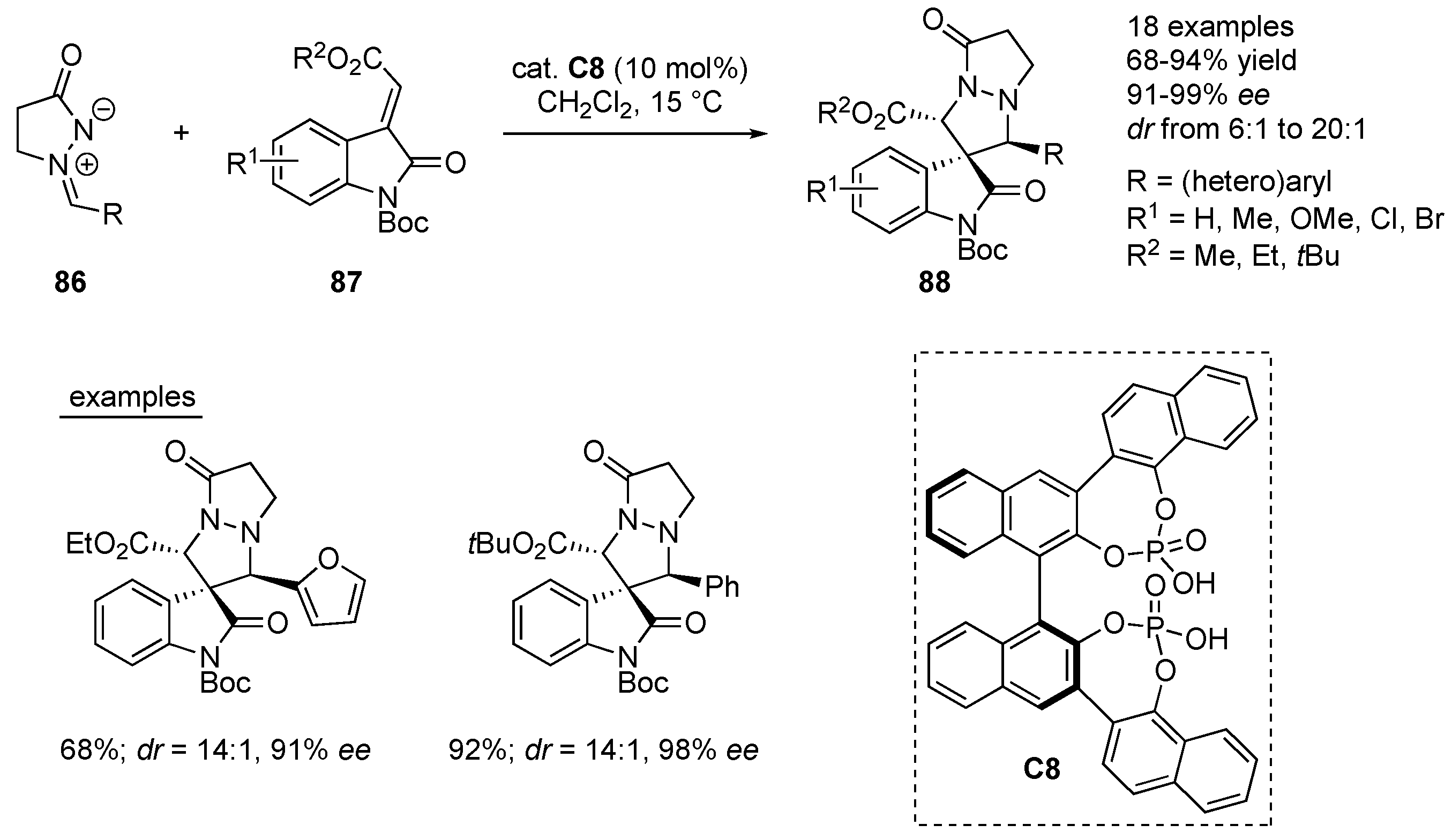
A chiral bis-phosphoric acid
C8, bearing triple axial chirality, was developed by Wang and co-workers and successfully utilized in the 1,3-dipolar cycloaddition of
N,
N-cyclic azomethine imines
86 to methyleneindolinones
87. The chiral spiro[pyrazolidin-3,3’-oxindole] products
88 have been formed in 68–94% yields, good to excellent diastereoselectivity (
dr from 6:1–20:1) and excellent enantioselectivity (91–99%
ee). Azomethine imines bearing different (hetero)aryl substituents, as well as different electronic nature, bulkiness or position of the substitution patterns on the methyleneindolinones had negligible effect on the efficiency and stereoselectivity of transformations under the optimized reaction conditions. DFT calculations have been performed to shed the light on the mechanism of the 1,3-dipolar cycloaddition (
Scheme 28) [
50].
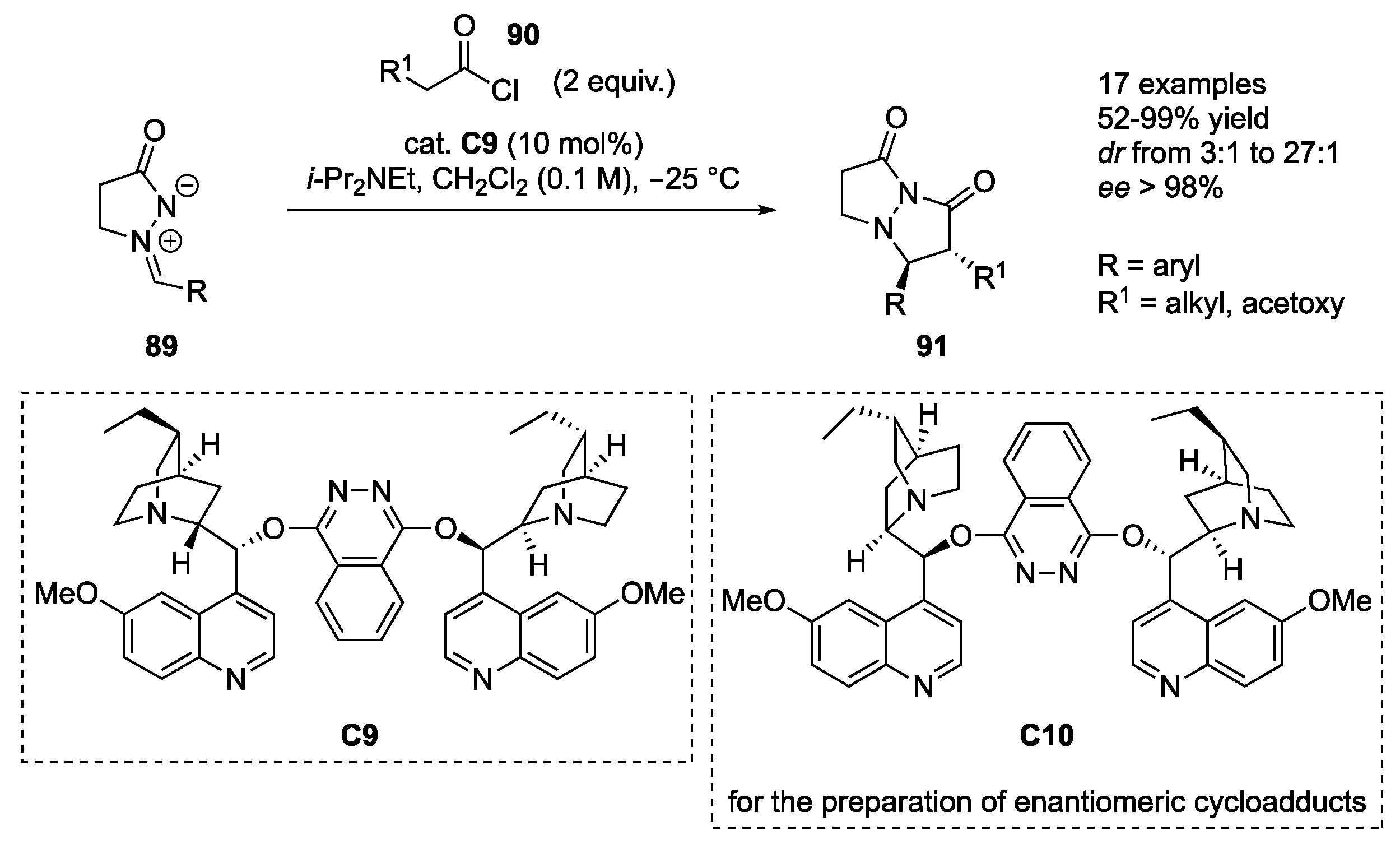
Kerrigan and co-workers reported 1,3-dipolar cycloadditions of the in situ-generated ketenes (formed from the corresponding acyl chlorides
90 and DIPEA) to azomethine imines
89 in the presence of quinuclidine organocatalyst
C9 as the catalyst of choice, thus furnishing (2
R,3
S)-bicyclic pyrazolidinones
91 in 52–99% yields, excellent enantioselectivity (≥96%
ee) and moderate to high diastereoselectivity (
dr 3:1–27:1). The method displays tolerance to aryl groups of azomethine imine containing both electron-donating and electron-withdrawing substituents, while with respect to ketene substituents, an acetoxy and different alkyl substituents were employed. With the acetoxy group, a markedly improved
trans-diastereoselectivity was observed (
dr 12:1 to 27:1). If pseudo enantiomeric catalyst
C10 was applied, enantiomeric (2
S,3
R)-bicyclic cycloadducts
91 were obtained, retaining the same level of stereoselectivity (
Scheme 29) [
51].
Proline-derived thiourea organocatalyst
C11 was used by Kesavan et al. for the formal [3+2] cycloaddition of azomethine imines
92 to malononitrile (
93), yielding bicyclic pyrazolidinones
94 with embedded enaminonitrile functionality in 68–92% yield and 41–98%
ee. Azomethine imines containing cyclohexyl, heteroaromatic and diversely substituted aromatic substituents were used. 2-methylmalononitrile also gave the expected product (78% yield, 70%
ee), while other dipolarophiles such as ethyl 2-cyanoacetate, benzoylacetonitrile and (phenylsulfonyl)acetonitrile failed to react. Presumably, the azomethine imine gets activated via hydrogen bonding with the thiourea moiety, whereas the tertiary amine deprotonates/activates the malononitrile (
Scheme 30) [
52].
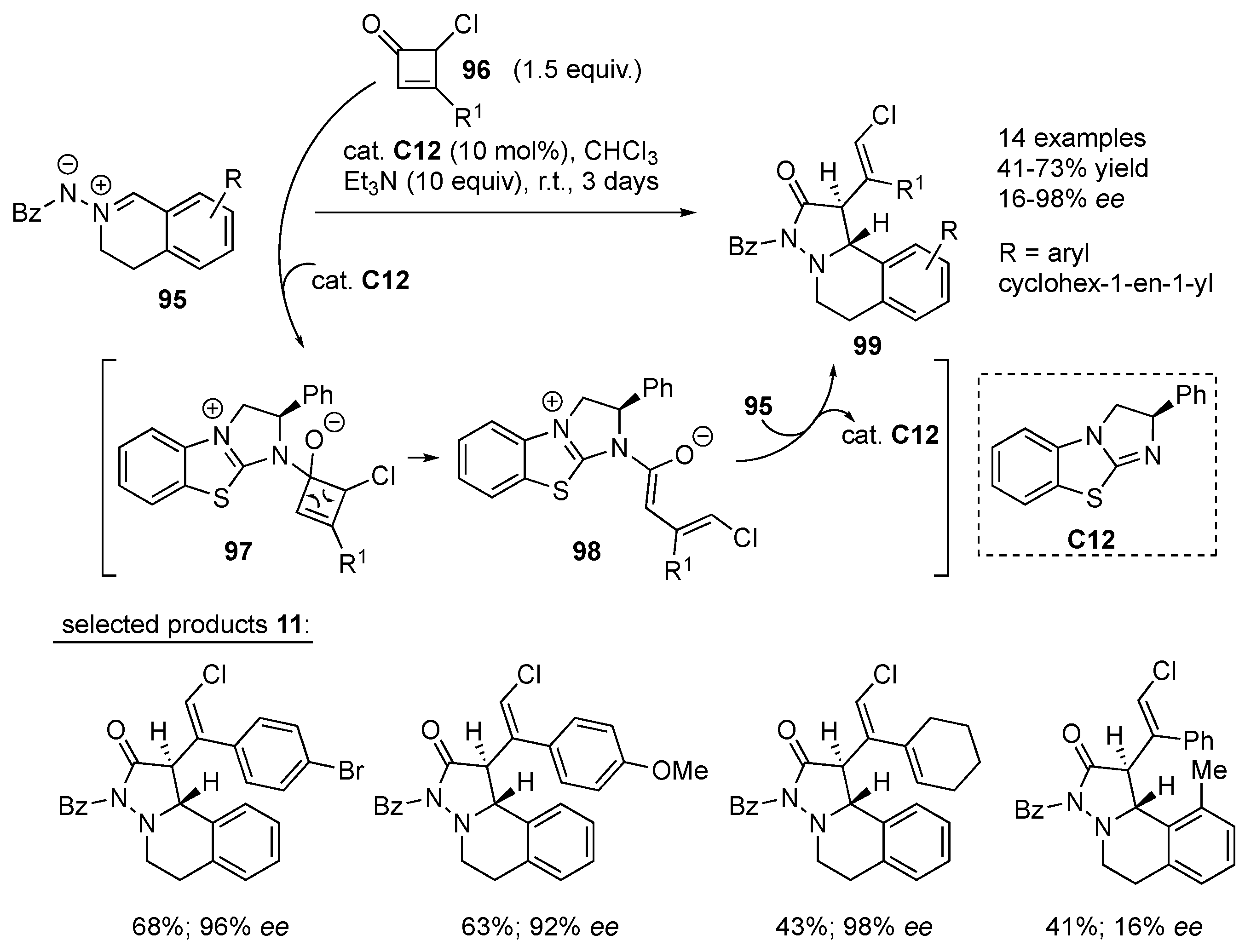
Chi and co-workers developed a new C–C bond activation of
γ-mono-chlorine-substituted cyclobutenones
96 enabled by an isothiourea Lewis base organocatalyst
C12. The 1,2-addition of Lewis base
C12 to
96 furnishes intermediate
97, which undergoes electrocyclic ring opening to generate vinyl enolate intermediate
98. Selective α-addition of
98 to azomethine imine
95 followed by cyclization gives the final products
28 in excellent diastereo- and enantioselectivity (
cis-selective, up to ≥ 98%
ee). The developed reaction tolerates different aromatic substituents at the β-carbon of cyclobutenone
96, while the mono-chlorine in the 3-position is essential for the positive outcome of the reaction. With regard to azomethine imine
95, substituents in position 8 other than hydrogen result in lower yields and enantiomeric excess, presumably due to steric hindrance (
Scheme 31) [
53].
Using chiral phosphine organocatalyst
C13, Shi et al. managed to employ
δ-substituted allenic esters
101 in an asymmetric [3+2] cycloaddition to
C,
N-cyclic azomethine imines
100, yielding tetrahydroquinoline derivatives
102 in 57–93% yields, with high diastereoselectivity (> 20:1
dr) and good enantioselectivity (68–93%
ee). The employed allenoates
101 react as C
2-synthons via their δ- and γ-positions, for which the authors proposed a plausible reaction mechanism. Both simple and sterically-demanding alkyl allenoates
101 have been successfully employed, containing benzyl and various aryl substituents in the δ-position, while the reaction with allenoate containing
δ-methyl-substituent failed. In the reaction of δ-Ph substituted allenoates
101 with various azomethine imines
100, the electron-rich alkyl-substituents on the azomethine imine seem to have a beneficial influence on the enantioselectivity of the reaction.
N,
N-cyclic azomethine imine does not react with δ-substituted allenic esters under the applied reaction conditions (
Scheme 32) [
54].
Ye and co-workers developed
N-heterocyclic carbene-catalyzed [3+2] cycloaddition of α-chloroaldehydes
104 to
C,
N-cyclic azomethine imines
103.
N-heterocyclic carbene was generated in situ from the corresponding triazolium salt
C14 and DIPEA. The corresponding pyrazolidinone products
105 were obtained in good yields (65–93%), with moderate to good diastereoselectivity (3:1–8:1
dr) and excellent enantioselectivity (90–99%
ee). It was found that both β-(hetero)aryl and alkyl α-chloroaldehydes
104, as well as azomethine imines
103 containing various
N-arylcarbonyl groups worked well, affording the corresponding products
105 with high enantioselectivities. On the other hand, the reaction of α-chlorophenylacetaldehyde with azomethine imine, under optimized reaction conditions, furnished the desired product in decreased yield (30%) and enantioselectivity (55%
ee), though significantly improved diastereoselectivity (
dr > 20:1) (
Scheme 33). A plausible catalytic cycle was postulated. It includes
N-heterocyclic carbene addition to α-chloroacetaldehyde, thus generating Breslow intermediate [
55], which decomposes to azolium enolate. Azolium enolate reacts with the azomethine imine in an addition-cyclization sequence, affording the final products
105 [
56].
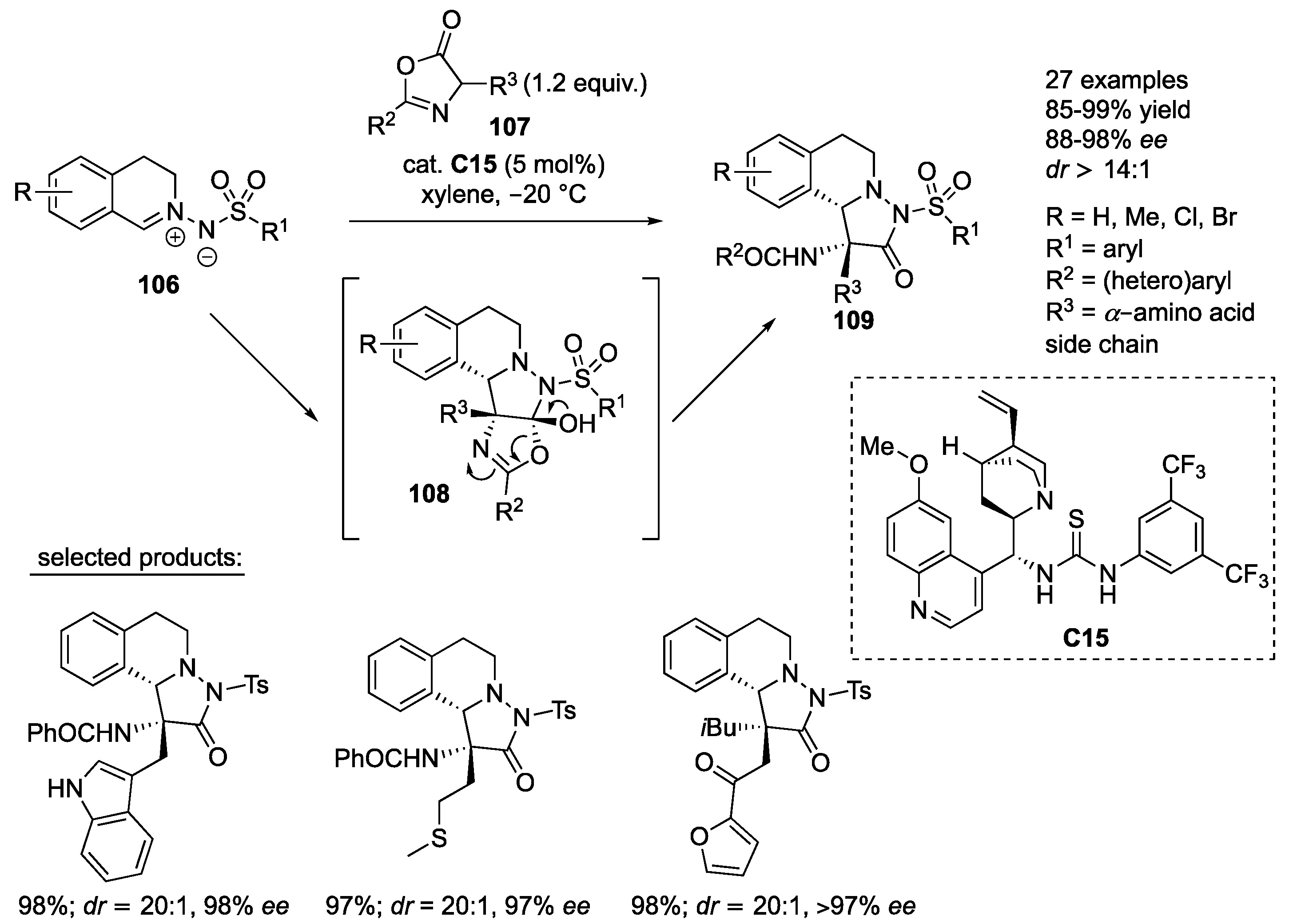
An organocatalyzed inverse electron demand 1,3-dipolar cycloaddition of
C,
N-cyclic azomethine imines
106 with azlactones
107 has been established by Su and co-workers employing noncovalent quinidine-derived bifunctional thiourea organocatalyst
C15. The initial tetracyclic intermediates
108 are unstable and rearrange into the final products
109. Products
109 were obtained in high yields (85–99%) with excellent stereoselectivity (88–98%
ee,
dr ≥ 14:1). In general, azlactones prepared from different amino acids were very well tolerated, as well as azlactones containing diverse (hetero)aryl substituents in the C2-position. Azomethine imines with different substituents on the aromatic ring and various substituents at the 4-position of the phenyl ring of the arylsulfonyl protecting group were very well compatible with the developed protocol (
Scheme 34) [
57].
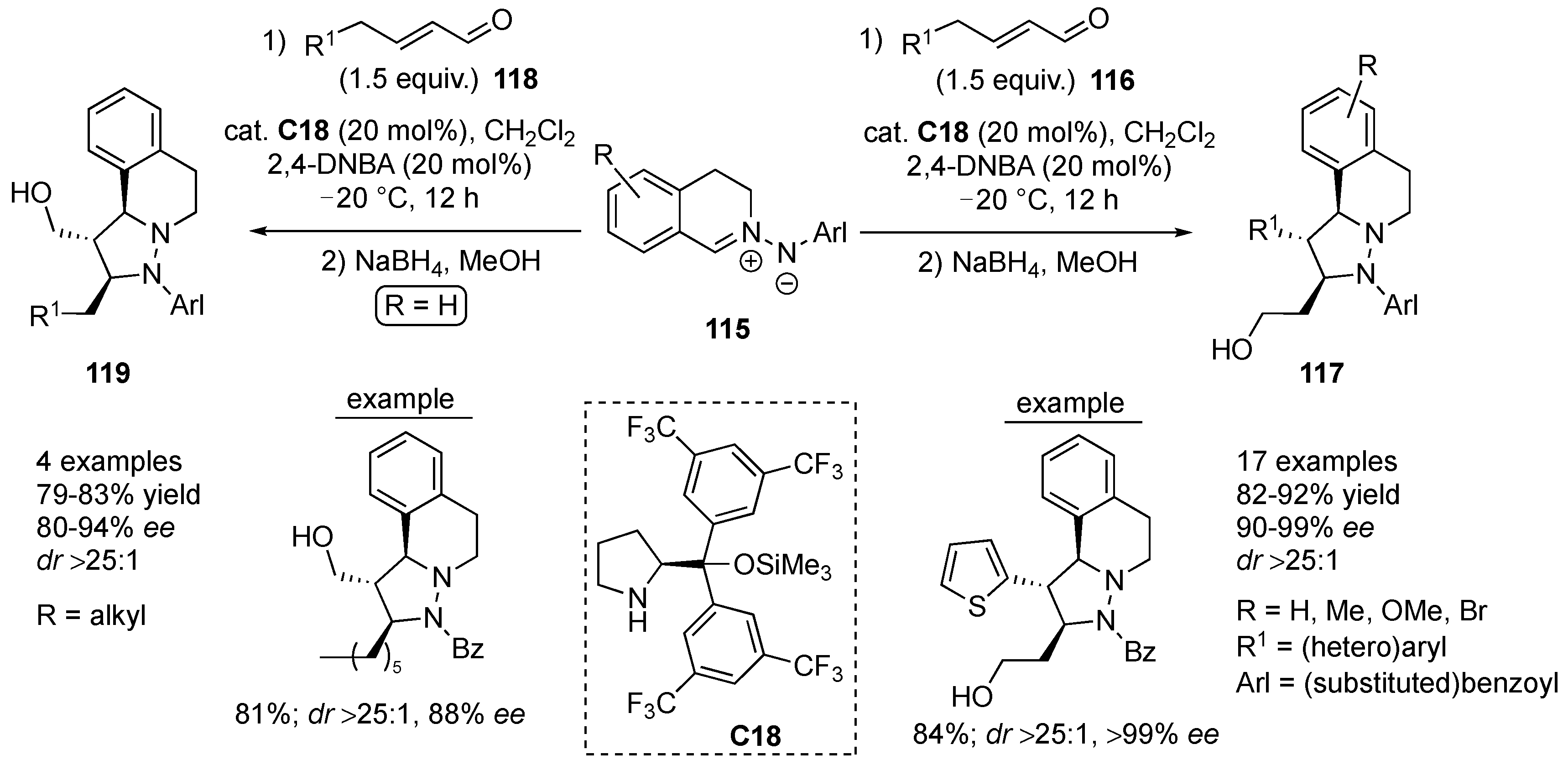
Zhu et al. developed an inverse electron demand cycloaddition between
C,
N-cyclic azomethine imines and electron-rich enecarbamates applying chiral phosphoric acids as organocatalysts. For the cycloaddition of benzyl
N-vinyl carbamate (
111) to azomethine imines
110 (containing both electron-donating and electron-withdrawing substituents, irrespective of their position on the aromatic ring), phosphoric acid
C16 turned out to be the optimal catalyst, yielding the corresponding products
112 in good to high yields (68–83%) with excellent regio- and stereo-selectivity (
dr ≥ 19:1, 88–98%
ee). On the other hand, cycloadditions to dipolarophiles
113 possessing an electron-rich (
Z)-internal bond needed re-optimization. Thus, the binol-based phosphoric acid
C17 was found to furnish the desired cycloadducts
114 in high yields (89–95%), excellent diastereoselectivity (
dr ≥ 19:1) and good to excellent enantioselectivity (80–98%
ee). Again, different substitution patterns (substituents) on the aromatic ring of azomethine imines
110 were well tolerated, whereas increasing the size of the β-substituent of the (
Z)-enecarbamate resulted in deceleration of the reaction, which at room temperature led to decreased enantioselectivity. Finally, reaction with benzyl [(
E)-prop-1-en-1-yl]carbamate afforded the corresponding product with significantly reduced stereoselectivity (
Scheme 35) [
58].
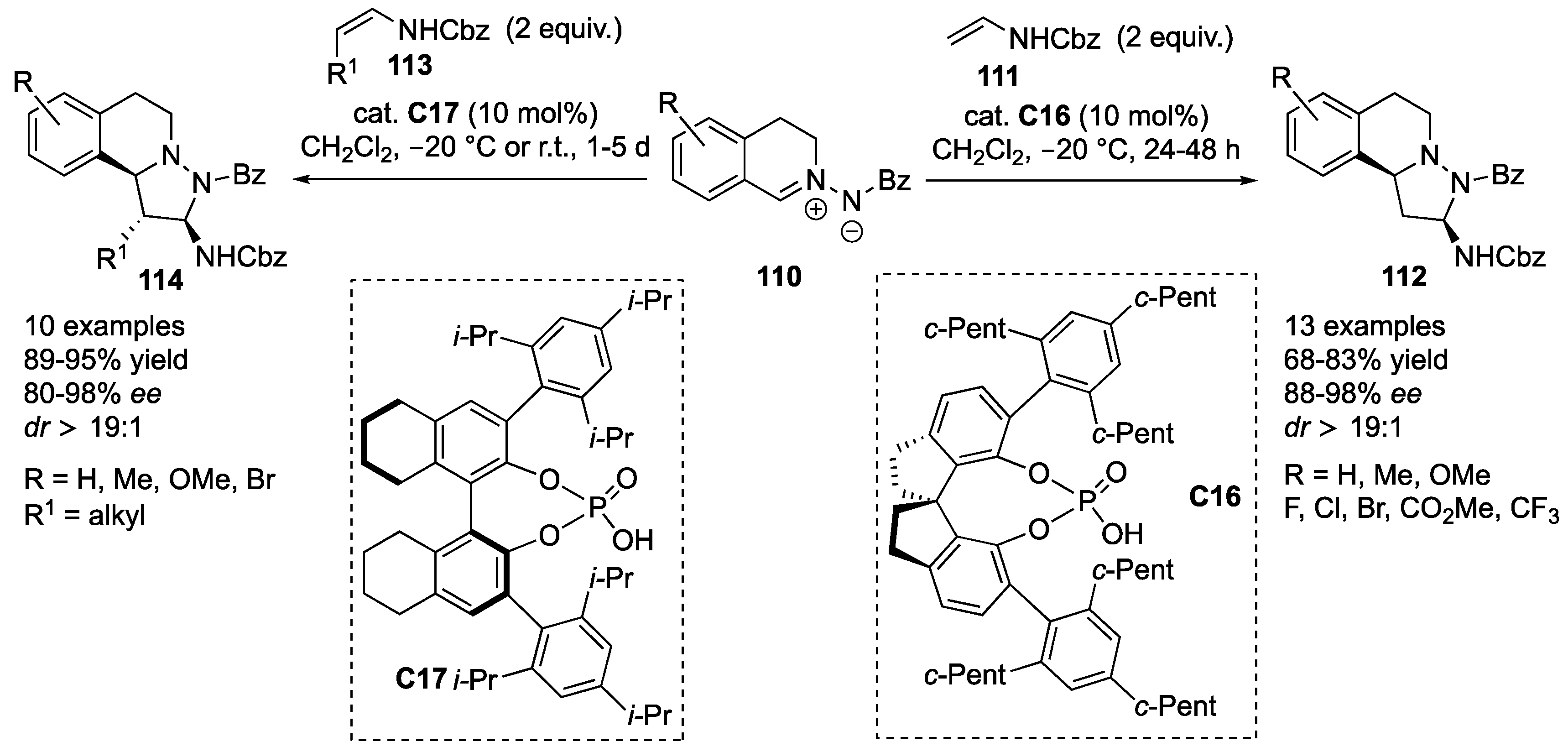
Wang and co-workers developed an asymmetric synthesis of tetrahydroquinoline derivatives via a [3+2] cycloaddition controlled by dienamine catalysis. Thus, the reaction of α,β-unsaturated aldehydes
116, containing (hetero)aromatic substituents in the γ-position, with
C,
N-cyclic azomethine imines
115 (either unsubstituted or containing 5-, 6- and 7-Me/MeO/Br substituents) catalyzed by chiral prolinol silyl ether
C18 [
59] furnished the desired tricyclic systems
117 in high yield (82–92%) with excellent stereoselectivity (
dr > 25:1, 90–99%
ee). This reaction proceeds via 4,5-reactivity of dienamine reactive intermediates. In stark contrast, reactions performed with aliphatic α,β-unsaturated aldehydes
116 with azomethine imines
115, performed under identical reaction conditions, yielded different [3+2]-cycloaddition products
119. In the latter case, the reaction proceeds via 3,4-reactivity of the iminium ion reactive intermediates. Products
119 were formed in 79–83% yields,
dr > 25:1 and 80–94%
ee (
Scheme 36) [
60].
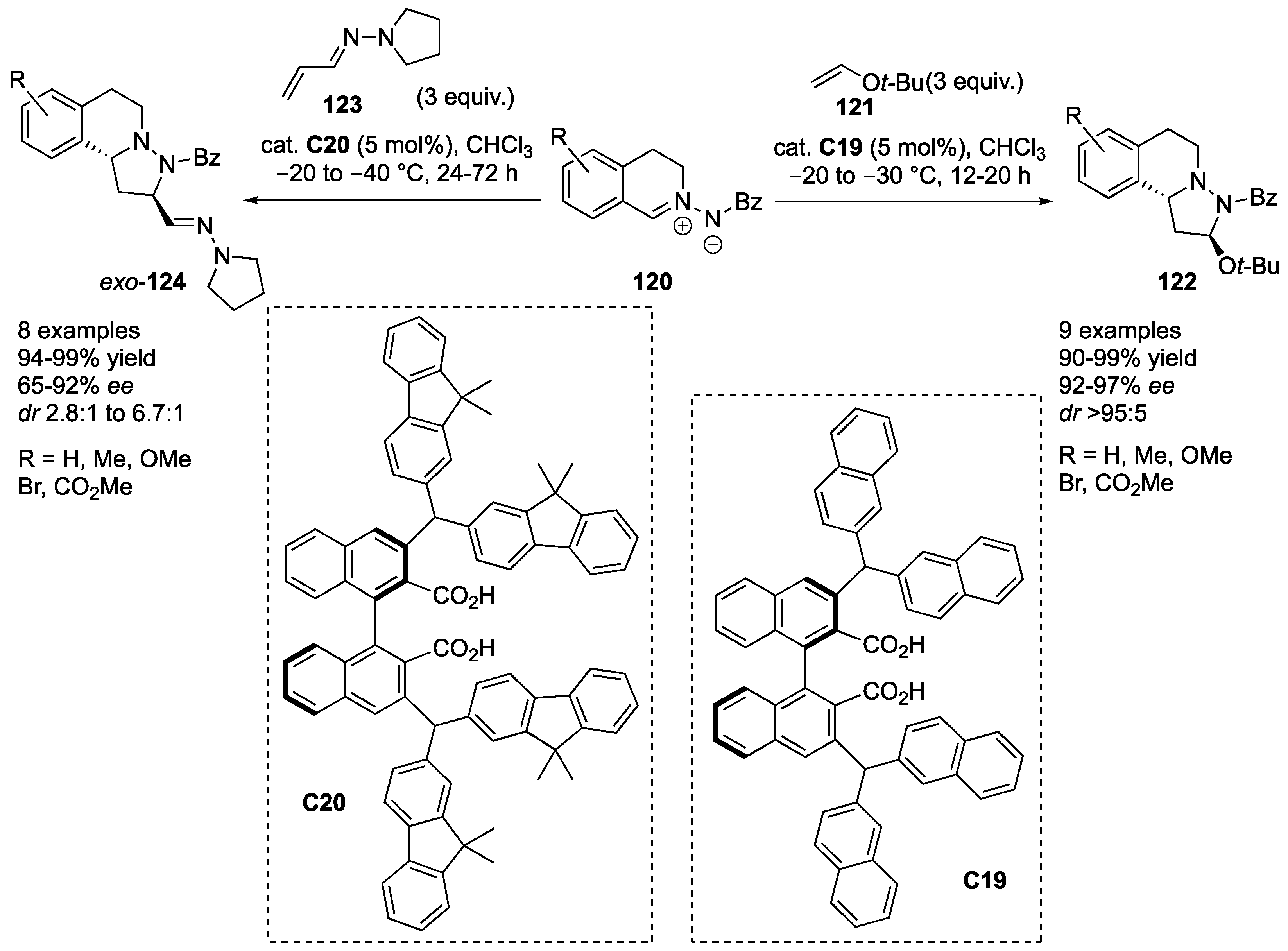
Maruoka and co-workers developed an organocatalyzed inverse electron demand cycloaddition of
C,
N-cyclic azomethine imines to electron-rich vinyl ether and vinylogous hydrazone catalyzed by axially chiral dicarboxylic acids. Thus, under optimized reaction conditions (organocatalyst
C19), azomethine imines
120, bearing either electron-rich or electron-deficient substituents, reacted with
tert-butyl vinyl ether (
121) to furnish the expected cycloadducts
122 in excellent yields (90–99%) and stereoselectivities (92–97%
ee,
exo/
endo > 95:5). Reactions of azomethine imines
120 with acrolein-derived vinylogous hydrazone
123 needed reoptimization of the reaction conditions (organocatalyst
C20). The corresponding cycloadducts
124 were formed in excellent yields (94–99%) and high enantioselectivity (65–92%
ee), albeit with modest diastereoselectivity (
exo/
endo = 2.8:1–6.7:1). The reaction with methacrolein-derived
α-substituted vinylogous hydrazone generated preferentially the
endo-diastereomer (
exo/
endo = 1.0:2.4) with moderate enantioselectivity (68%
ee), while β-substituted hydrazone derived from crotonaldehyde failed to give the desired product (
Scheme 37) [
61].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






































