Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
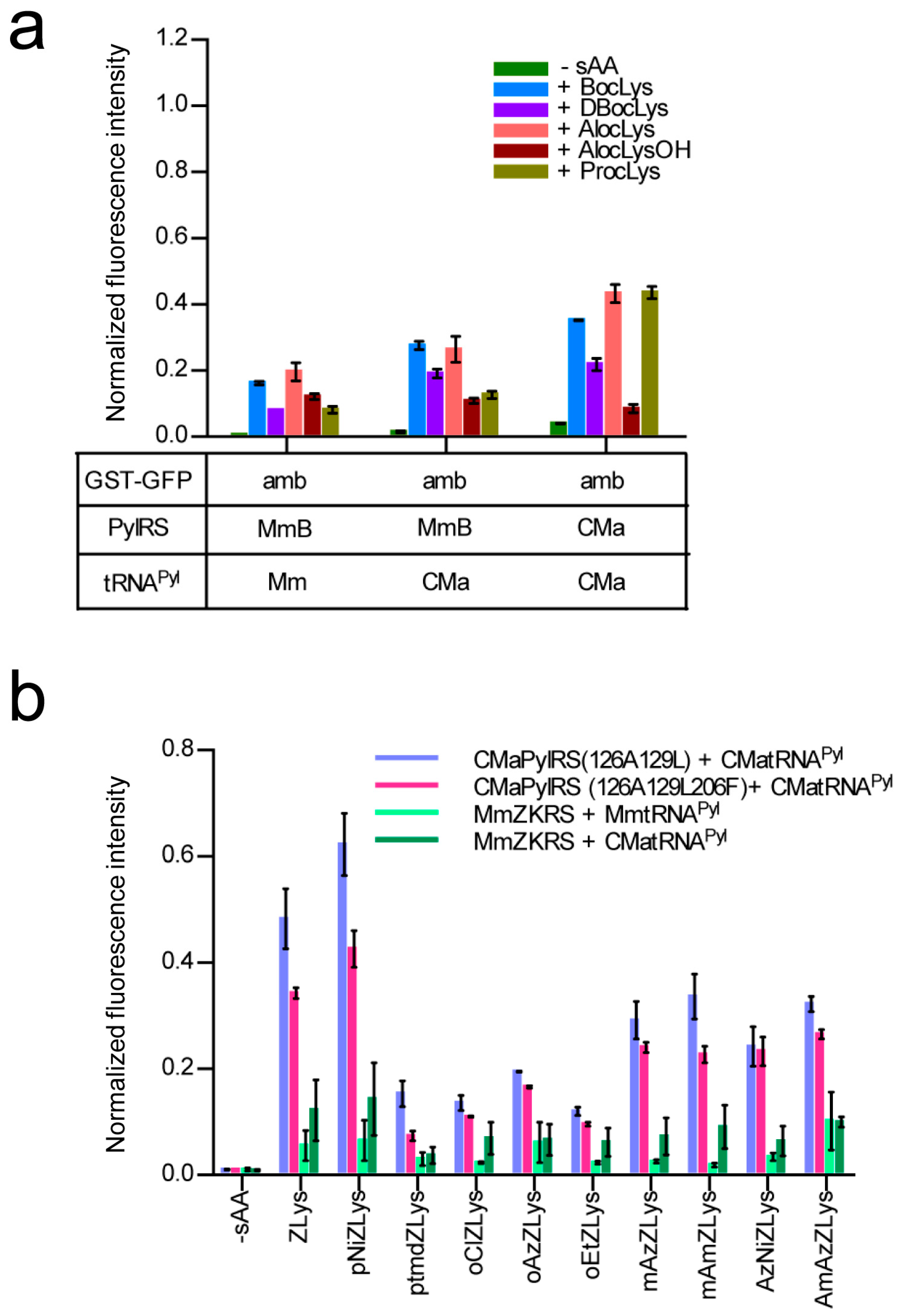
2.1. E. coli Cell-Based Incorporation of Lysine Derivatives Using the Wild-Type Ca. M. alvus PylRS
2.2. Mass Spectromeric Analysis to Identify the Incorporated Amino Acid
2.3. E. coli Cell-Free Incorporation of a Lysine Derivative using the Wild-Type Ca. M. alvus PylRS
2.4. Activities of Ca. M. alvus tRNAPyl Variants in E. coli Cell-Based Translation
2.5. E. coli Cell-Based Incorporation of Various Lysine Derivatives using the Wildtype and Variant CMaPylRS Molecules
3. Discussion
4. Materials and Methods
4.1. Amino Acids
4.2. Plasmids
4.3. Fluorescence Measurements of the E. coli Cell Culture
4.4. Mass Spectrometry
4.5. In Vitro Synthesis of tRNAPyl
4.6. Purification of PylRS
4.7. Cell-Free Translation
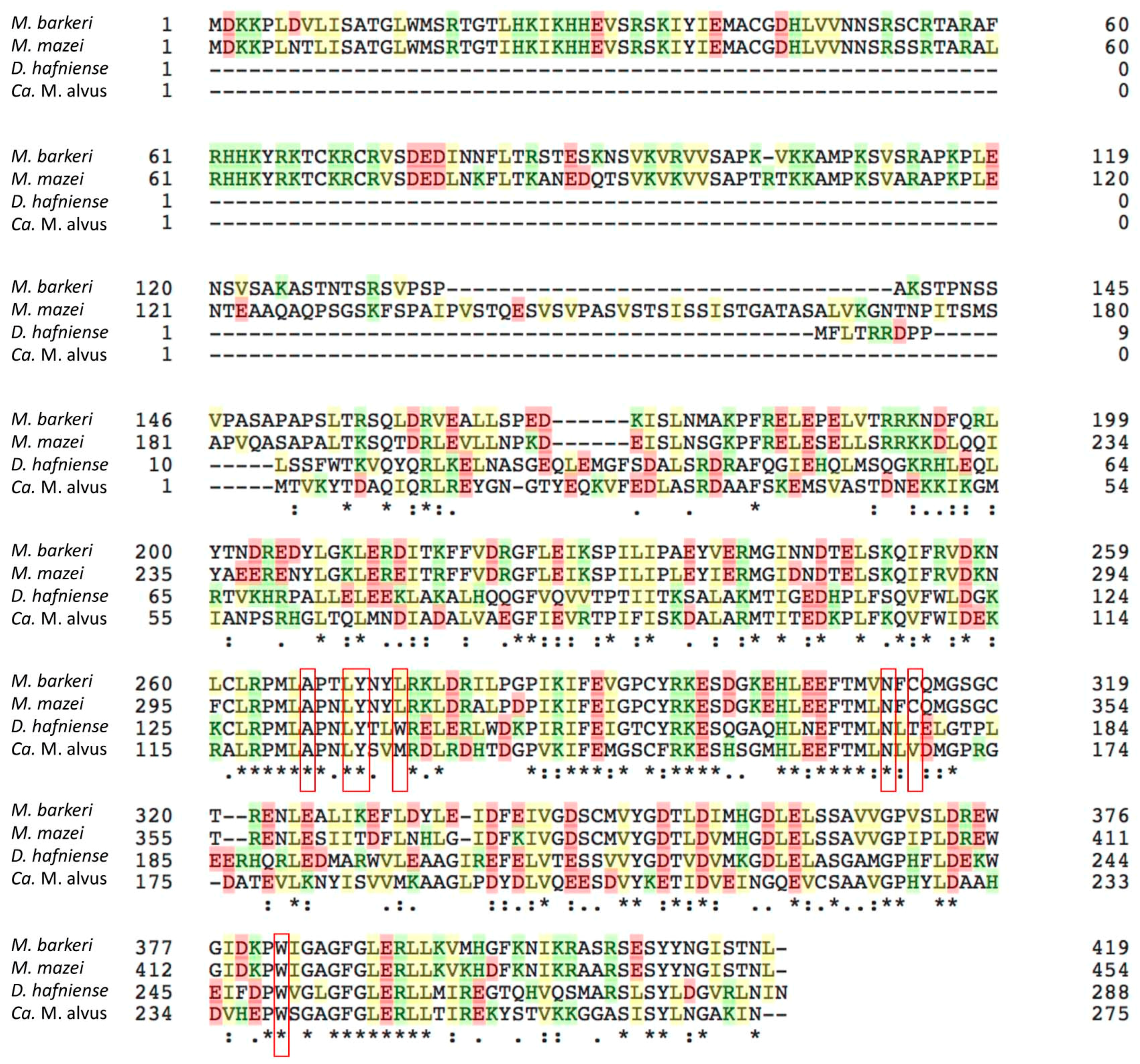
4.8. Sequence Alignment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gaston, M.A.; Jiang, R.; Krzycki, J.A. Functional context, biosynthesis, and genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 2011, 14, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann-Staubitz, P.; Neumann, H. The use of unnatural amino acids to study and engineer protein function. Curr. Opin. Struct. Biol. 2016, 38, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Crnković, A.; Suzuki, T.; Söll, D.; Reynolds, N.M. Pyrrolysyl-tRNA synthetase, an aminoacyl-tRNA synthetase for genetic code expansion. Croat. Chem. Acta 2016, 89, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Crnković, A.; Umehara, T.; Ivanova, N.N.; Kyrpides, N.C.; Söll, D. RNA-dependent cysteine biosynthesis in bacteria and archaea. MBio 2017, 8, e00561-17. [Google Scholar] [CrossRef] [PubMed]
- Tharp, J.M.; Ehnbom, A.; Liu, W.R. tRNAPyl: Structure, function, and applications. RNA Biol. 2018, 15, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Ambrogelly, A.; Gundllapalli, S.; Herring, S.; Polycarpo, C.; Frauer, C.; Söll, D. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc. Natl. Acad. Sci. USA 2007, 104, 3141–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnakumar, R.; Prat, L.; Aerni, H.; Ling, J.; Merryman, C.; Glass, J.I.; Rinehart, J.; Söll, D. Transfer RNA misidentification scrambles sense codon recoding. ChemBioChem 2013, 14, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Wang, W.; Liu, W.R. Towards Reassigning the Rare AGG Codon in Escherichia coli. Chembiochem 2014, 15, 1750–1754. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Recognition of non-α-amino substrates by pyrrolysyl-tRNA synthetase. J. Mol. Biol. 2009, 385, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Yang, M.Y.; Huang, Y.C.; Li, Y.T.; Chen, P.R.; Liu, L. Ligation of expressed protein α-hydrazides via genetic incorporation of an α-hydroxy acid. ACS Chem. Biol. 2012, 15, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Bindman, N.A.; Bobeica, S.C.; Liu, W.R.; van der Donk, W.A. Facile removal of leader peptides from lanthipeptides by incorporation of a hydroxy acid. J. Am. Chem. Soc. 2015, 137, 6975–6978. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, K.; Mukai, T.; Iraha, F.; Takahashi, M.; Haruna, K.; Date, M.; Yokoyama, K.; Sakamoto, K. Engineering an auto-maturing transglutaminase with enhanced thermostability by genetic code expansion with two codon reassignments. ACS Synth. Biol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Herring, S.; Ambrogelly, A.; Gundllapalli, S.; O’Donoghue, P.; Polycarpo, C.R.; Söll, D. The amino-terminal domain of pyrrolysyl-tRNA synthetase is dispensable in vitro but required for in vivo activity. FEBS Lett. 2007, 581, 3197–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Krzycki, J.A. PylSn and the homologous N-terminal domain of pyrrolysyl-tRNA synthetase bind the tRNA that is essential for the genetic encoding of pyrrolysine. J. Biol. Chem. 2012, 287, 32738–32746. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Nureki, O.; Yokoyama, S. Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of pyrrolysyl-tRNA synthetase from the methanogenic archaeon Methanosarcina mazei. Acta Crystallogr. Sect F Struct. Biol. Cryst. Commun. 2006, 62, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Sun, S.B.; Furman, J.L.; Xiao, H.; Schultz, P.G. A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 2013, 52, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Harris, H.M.; Tottey, W.; Mihajlovski, A.; Parisot, N.; Peyretaillade, E.; Peyret, P.; Gribaldo, S.; O’Toole, P.W.; Brugère, J.F. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 2012, 194, 6944–6945. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Gaci, N.; Peyret, P.; O’Toole, P.W.; Gribaldo, S.; Brugère, J.F. Unique characteristics of the pyrrolysine system in the 7th order of methanogens: Implications for the evolution of a genetic code expansion cassette. Archaea 2014, 2014, 374146. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Parisot, N.; Harris, H.M.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genom. 2014, 15, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, J.C.W.; Chin, J.W. Mutually orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs. Nat. Chem. 2018, 10, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; O’Donoghue, P.; Gundllapalli, S.; Araiso, Y.; Ishitani, R.; Umehara, T.; Söll, D.; Nureki, O. Pyrrolysyl-tRNA synthetase-tRNA(Pyl) structure reveals the molecular basis of orthogonality. Nature 2009, 457, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(epsilon)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Kobayashi, T.; Hino, N.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem. Biophys. Res. Commun. 2008, 371, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Hino, H.; Iraha, F.; Mukai, T.; Sakamoto, K.; Yokoyama, S. Wide-range protein photo-crosslinking achieved by a genetically encoded Nε-(benzyloxycarbonyl)lysine derivative with a diazirinyl moiety. Mol. BioSyst. 2012, 8, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Matsuda, T.; Ohtake, K.; Yanagisawa, T.; Yokoyama, S.; Fujiwara, Y.; Watanabe, T.; Hohsaka, T.; Sakamoto, K. Incorporation of a doubly functionalized synthetic amino acid into proteins for creating chemical and light-induced conjugates. Bioconjug. Chem. 2016, 27, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Kavran, J.M.; Gundllapalli, S.; O’Donoghue, P.; Englert, M.; Söll, D.; Steitz, T.A. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Crystallographic studies on multiple conformational states of active-site loops in pyrrolysyl-tRNA synthetase. J. Mol. Biol. 2008, 378, 634–652. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell. 2009, 36, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Hahn, L.E.; Heitmüller, S.; Frauendorf, H.; Valerius, O.; Braus, G.H.; Neumann, H. Genetically encoding lysine modifications on histone H4. ACS Chem. Biol. 2015, 10, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Xuan, W.; Shao, S.; Liu, T.; Schultz, P.G. Genetic incorporation of ε-N-2-hydroxyisobutyryl-lysine into recombinant histones. ACS Chem. Biol. 2015, 10, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.A.; Cropp, T.A. Genetic encoding of the post-translational modification 2-hydroxyisobutyryl-lysine. Org. Biomol. Chem. 2015, 13, 6479–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The described plasmids are available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, A.; Iraha, F.; Ohtake, K.; Sakamoto, K. Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes. Molecules 2018, 23, 2460. https://doi.org/10.3390/molecules23102460
Yamaguchi A, Iraha F, Ohtake K, Sakamoto K. Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes. Molecules. 2018; 23(10):2460. https://doi.org/10.3390/molecules23102460
Chicago/Turabian StyleYamaguchi, Atsushi, Fumie Iraha, Kazumasa Ohtake, and Kensaku Sakamoto. 2018. "Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes" Molecules 23, no. 10: 2460. https://doi.org/10.3390/molecules23102460
APA StyleYamaguchi, A., Iraha, F., Ohtake, K., & Sakamoto, K. (2018). Pyrrolysyl-tRNA Synthetase with a Unique Architecture Enhances the Availability of Lysine Derivatives in Synthetic Genetic Codes. Molecules, 23(10), 2460. https://doi.org/10.3390/molecules23102460