Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities

Abstract
:
1. Introduction
2. Results
3. Materials and Methods
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compounds 1 and 2 to Determine the Absolute Configuration of the Monosaccharides
3.5. Anti-Inflammatory Activity Assay Against NO Production
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, H. Search the origin of Araceae by ecological geography. Acta Bot. Yunnanica 1996, 18, 14–42. [Google Scholar]
- El-Desouky, S.K.; Kim, K.H.; Ryu, S.Y.; Eweas, A.F.; Gamal-Eldeen, A.M.; Kim, Y.K. A new pyrrole alkaloid isolated from Arum palaestinum Boiss. and its biological activities. Arch. Pharm. Res. 2007, 30, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Yi, X.M.; Feng, J.Y.; Wang, Y.H.; He, X.J. Piperidine alkaloids from Alocasiamacrorrhiza. Phytochemistry 2017, 143, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.R.; Bliss, B.J.; Suzuki, J.Y.; Borris, R.P. Chemotaxonomy of Hawaiian Anthurium cultivars based on multivariate analysis of phenolic metabolites. J. Agric. Food Chem. 2014, 62, 11323–11334. [Google Scholar] [CrossRef] [PubMed]
- Roshan, R.; Ahmed, S.; Hasan, M.M. Arisaema jacquemontii Blume (Araceae): A review of medicinal uses, phytochemistry and pharmacology. J. Pharmacogn. Phytochem. 2017, 6, 429–432. [Google Scholar]
- Wu, Y.Y.; Huang, X.X.; Zhang, M.Y.; Zhou, L.; Li, D.Q.; Cheng, Z.Y.; Li, L.Z.; Peng, Y.; Song, S.J. Chemical constituents from the tubers of Pinellia ternate (Araceae) and their chemotaxonomic interest. Biochem. Syst. Ecol. 2015, 62, 236–240. [Google Scholar] [CrossRef]
- Li, D.D.; Fang, M.L.; Liu, Q.; Li, Q.L.; Gao, Y.T. Study on the extraction of polyphenol from Scindapsus officinalis with ultrasonic wave technology optimized by central composite design-response surface method. J. Chin. Mater. Med. 2011, 34, 129–133. [Google Scholar]
- Yu, J.Q.; Song, X.Y.; Wang, D.J.; Wang, X.Y.; Wang, X. Five new chromone glycosides from Scindapsus officinalis (Roxb.) Schott. Fitoterapia 2017, 122, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Q.; Song, X.Y.; Yang, P.; Wang, X.Y.; Wang, X. Alkaloids from Scindapsus officinalis (Roxb.) Schott. and their biological activities. Fitoterapia 2018, 129, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Jia, X.C.; Luo, Q.W.; Zhang, Q.; Luo, B.; Liu, W.B.; Zhang, X.; Xu, Q.L.; Tan, J.W. Phenolics fromMikaniamicrantha and their antioxidant activity. Molecules 2017, 22, 1140. [Google Scholar] [CrossRef] [PubMed]
- Kurashima, K.; Fujii, M.; Ida, Y.; Akita, H. Simple synthesis of β-d-Glycopyranosides using β-Glycosidase from almonds. Chem. Pharm. Bull. 2004, 52, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.C.; Wang, Y.; Yu, C.M.; Yu, D.H.; Chen, P.P.; Liu, S.M. Two new saccharide fatty acid esters from the fruit of Morindacitrifolia L. and their ABTS radical scavenging activities. Rec. Nat. Prod. 2014, 8, 25–31. [Google Scholar]
- Marzouk, M.S.; Moharram, F.A.; El Dib, R.A.; El-Shenawy, S.M.; Tawfike, A.F. Polyphenolic profile and bioactivity study of Oenotheraspeciosa Nutt. aerial parts. Molecules 2009, 14, 1456–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.Y.; Hu, L.H. Study on the chemical constituents of Daphniphyllum angustifolium. Helv. Chim. Acta 2006, 89, 884–894. [Google Scholar] [CrossRef]
- Wang, H.B.; Nair, M.G.; Strasbur, G.M.; Booren, A.M.; Gray, J.I. Novel antioxidant compounds from Tart Cherries (Prunuscerasus). J. Nat. Prod. 1999, 62, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Syntheses of optically active grasshopper ketone and dehydrovomifoliol as a synthetic support for the revised absolute configuration of (+)-abscisic acid. Tetrahedron 1974, 30, 1065–1072. [Google Scholar] [CrossRef]
- Della, G.M.; Marino, C.D.; Zarrelli, A. Isolation and phytotoxicity of apocarotenoids from Chenopodium album. J. Nat. Prod. 2004, 67, 1492–1495. [Google Scholar]
- Chae, H.S.; Yoo, H.; Kim, Y.M.; Choi, Y.H.; Lee, C.H.; Chin, Y.W. Anti-Inflammatory Effects of 6,8-Diprenyl-7,4′-dihydroxyflavanone from Sophoratonkinensis on Lipopolysaccharide-Stimulated RAW 264.7 Cells. Molecules 2016, 21, 1049. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (1–3) are available from the authors. |

 C) correlations of 1–3 (left), and key NOESY (H
C) correlations of 1–3 (left), and key NOESY (H  H) correlations of 1 and 3 (right).
C) correlations of 1–3 (left), and key NOESY (H H) correlations of 1 and 3 (right).
H) correlations of 1 and 3 (right).
C) correlations of 1–3 (left), and key NOESY (H H) correlations of 1 and 3 (right).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Positions | 1 a | 2 b | 3 b | |||
|---|---|---|---|---|---|---|
| 1 | 79.4 | 144.86 | 5.10 dd (4.8,6.6) | 70.2 | ||
| 2 | 5.98 dd (11.2, 17.6) | 144.1 | 144.87 | 3.78 dd (4.2, 6.6) | 68.0 | |
| 3 | 5.09 overlapped | 114.1 | 6.80 d (1.8) | 112.9 | 127.4 | |
| 4a | 1.96 m | 22.6 | 133.4 | 2.65–2.69 m | 27.9 | |
| 4b | 2.21–2.25 m | |||||
| 5 | 1.50 m | 40.2 | 6.67 dd (1.8, 8.4) | 120.8 | 4.28 m | 65.8 |
| 6 | 5.09 overlapped | 125.3 | 7.00 d (8.4) | 115.53 | 6.72 brs | 140.1 |
| 7 | 130.8 | 3.30 d (6.6) | 39.1 | 166.7 | ||
| 8 | 1.61 s | 25.9 | 5.94 m | 137.9 | ||
| 9 | 1.54 s | 18.0 | 5.03–5.08 m | 115.49 | ||
| 10 | 1.22 s | 24.0 | ||||
| 1′ | 4.13 d (8.0) | 98.2 | 4.84 d (7.2) | 100.2 | 125.9 | |
| 2′ | 2.90 m | 74.0 | 3.23–3.28 m | 73.1 | 7.05 d (2.0) | 115.3 |
| 3′ | 3.09 t (8.4) | 77.5 | 3.23–3.28 m | 76.98 | 146.0 | |
| 4′ | 3.02 t (9.2) | 70.7 | 3.23–3.28 m | 76.78 | 148.9 | |
| 5′ | 2.95 m | 77.1 | 3.13–3.17 m | 69.7 | 6.76 d (8.4) | 116.2 |
| 6′a | 3.58 d (11.2) | 61.7 | 3.65–3.67 m | 60.8 | 7.01 dd (2.0, 8.4) | 121.9 |
| 6′b | 3.38 m | 3.42–3.45 m | ||||
| 7′ | 7.48 d (15.6) | 145.9 | ||||
| 8′ | 6.25 d (15.6) | 114.4 | ||||
| 9′ | 166.5 | |||||
| 1″ | 4.09 d (7.8) | 102.8 | ||||
| 2″ | 2.93 td (8.4, 4.2) | 73.4 | ||||
| 3″ | 3.05–3.08 m | 76.85 | ||||
| 4″ | 3.10–3.13 m | 76.81 | ||||
| 5″ | 3.01–3.05 m | 70.1 | ||||
| 6″a | 3.65–3.67 m | 61.1 | ||||
| 6″b | 3.42–3.45 m | |||||
| 1‴a | 3.74 d (13.2) | 68.5 | ||||
| 1‴b | 3.40 d (13.2) | |||||
| 2‴ | 31.2 | |||||
| 3‴ | 1.23–1.32 ov | 25.2 | ||||
| 4‴a | 1.49–1.53 m | 29.2 | ||||
| 4‴b | 1.23–1.32 ov | |||||
| 5‴ | 1.23–1.32 ov | 21.9 | ||||
| 6‴ | 0.87 t (7.2) | 13.9 | ||||
| OMe | 3.74 s (OMe-2) | 55.6 | 3.69 s | 52.3 |
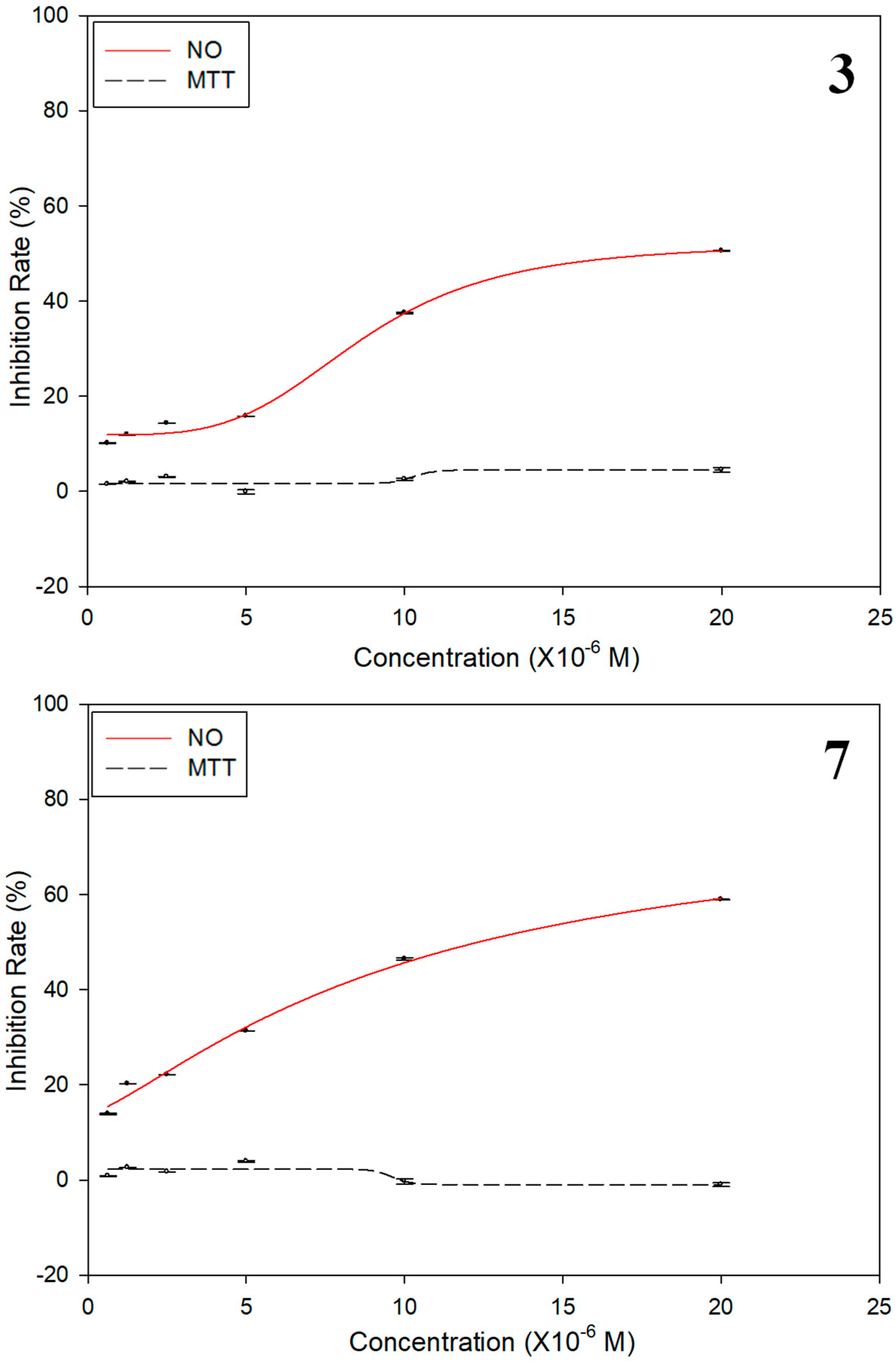
| Compounds | IC50 (μM) | Inhibition Rate (%) b |
|---|---|---|
| NO | Cytotoxicity | |
| 1 | >100 | 1.8 |
| 2 | >100 | −0.5 |
| 3 | 12.2 ± 0.8 | 4.7 |
| 4 | >100 | 1.1 |
| 5 | >100 | 4.5 |
| 6 | >100 | 1.2 |
| 7 | 18.9 ± 0.3 | 2.4 |
| Dexamethasone | 2.2 ± 0.02 | NT c |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Geng, Y.; Wang, X.; Song, X.; Wang, X.; Yu, J. Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules 2018, 23, 2577. https://doi.org/10.3390/molecules23102577
Dong H, Geng Y, Wang X, Song X, Wang X, Yu J. Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules. 2018; 23(10):2577. https://doi.org/10.3390/molecules23102577
Chicago/Turabian StyleDong, Hongjing, Yanling Geng, Xueyong Wang, Xiangyun Song, Xiao Wang, and Jinqian Yu. 2018. "Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities" Molecules 23, no. 10: 2577. https://doi.org/10.3390/molecules23102577
APA StyleDong, H., Geng, Y., Wang, X., Song, X., Wang, X., & Yu, J. (2018). Chemical Constituents from Scindapsus officinalis (Roxb.) Schott. and Their Anti–Inflammatory Activities. Molecules, 23(10), 2577. https://doi.org/10.3390/molecules23102577