Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Results
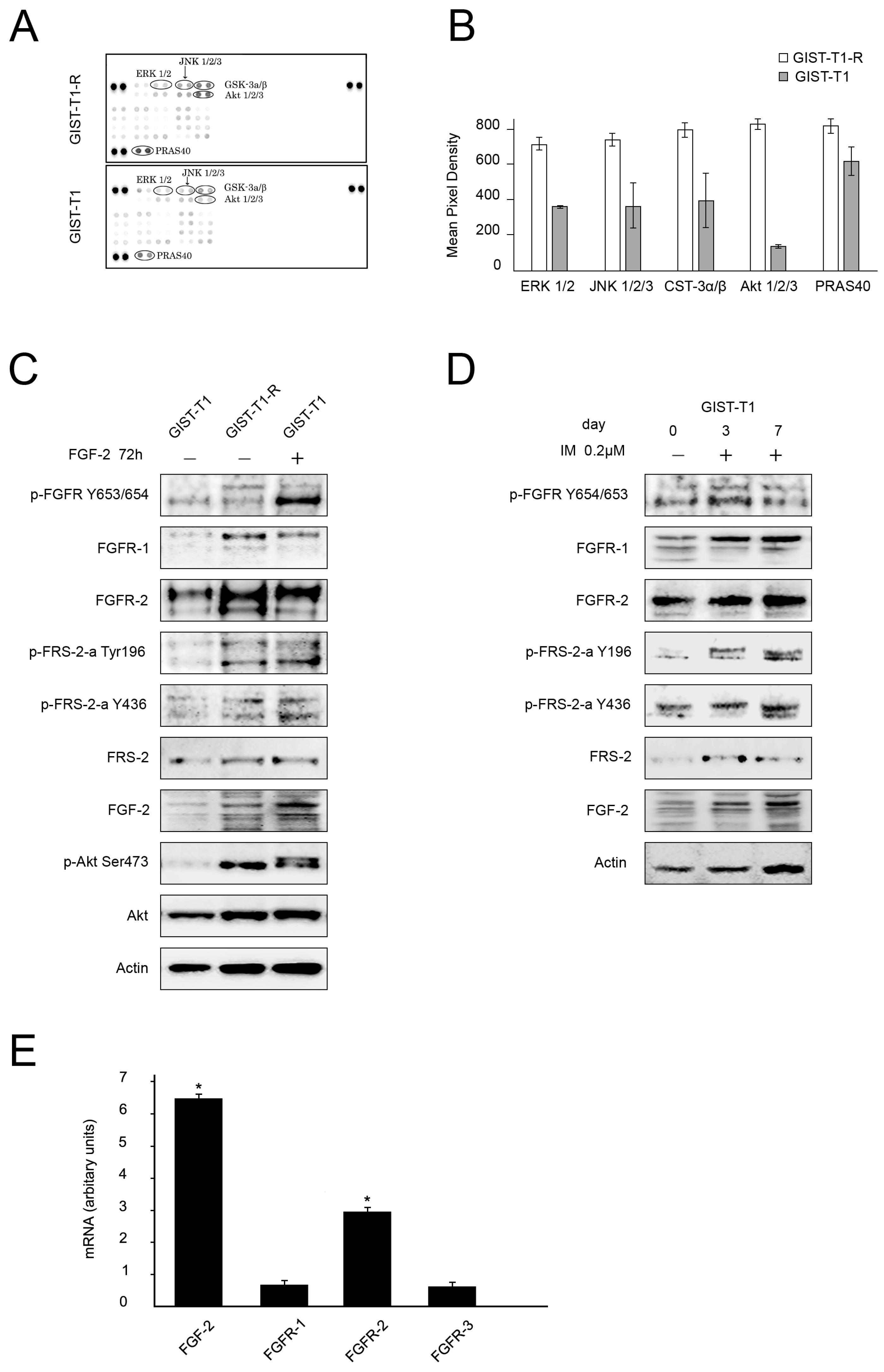
2.1. FGF/FGFR Signaling Is Activated in IM-Resistant GIST Cells
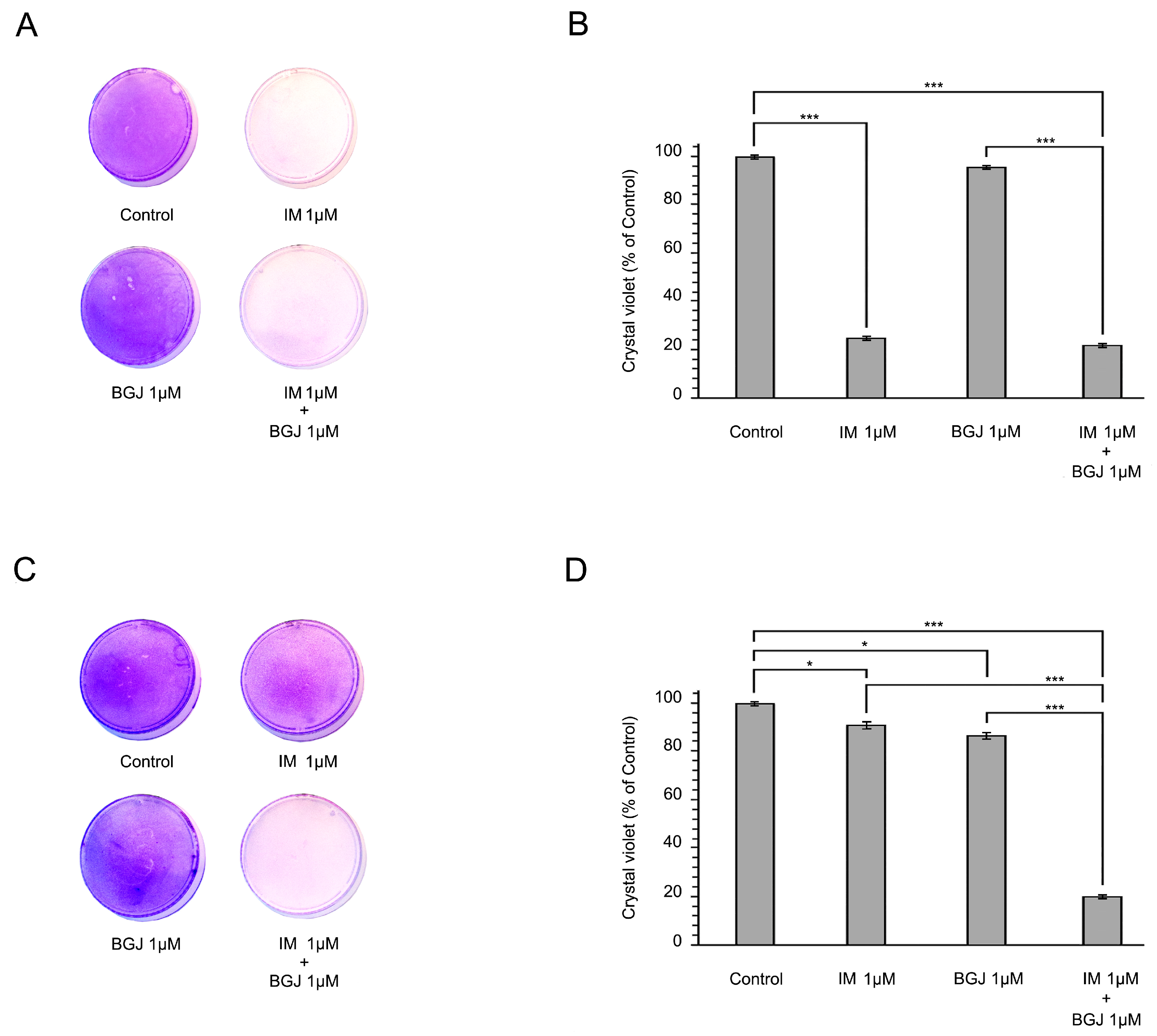
2.2. BGJ398 Restores the Growth Inhibitory Effect of IM in GIST T1-R Cells In Vitro
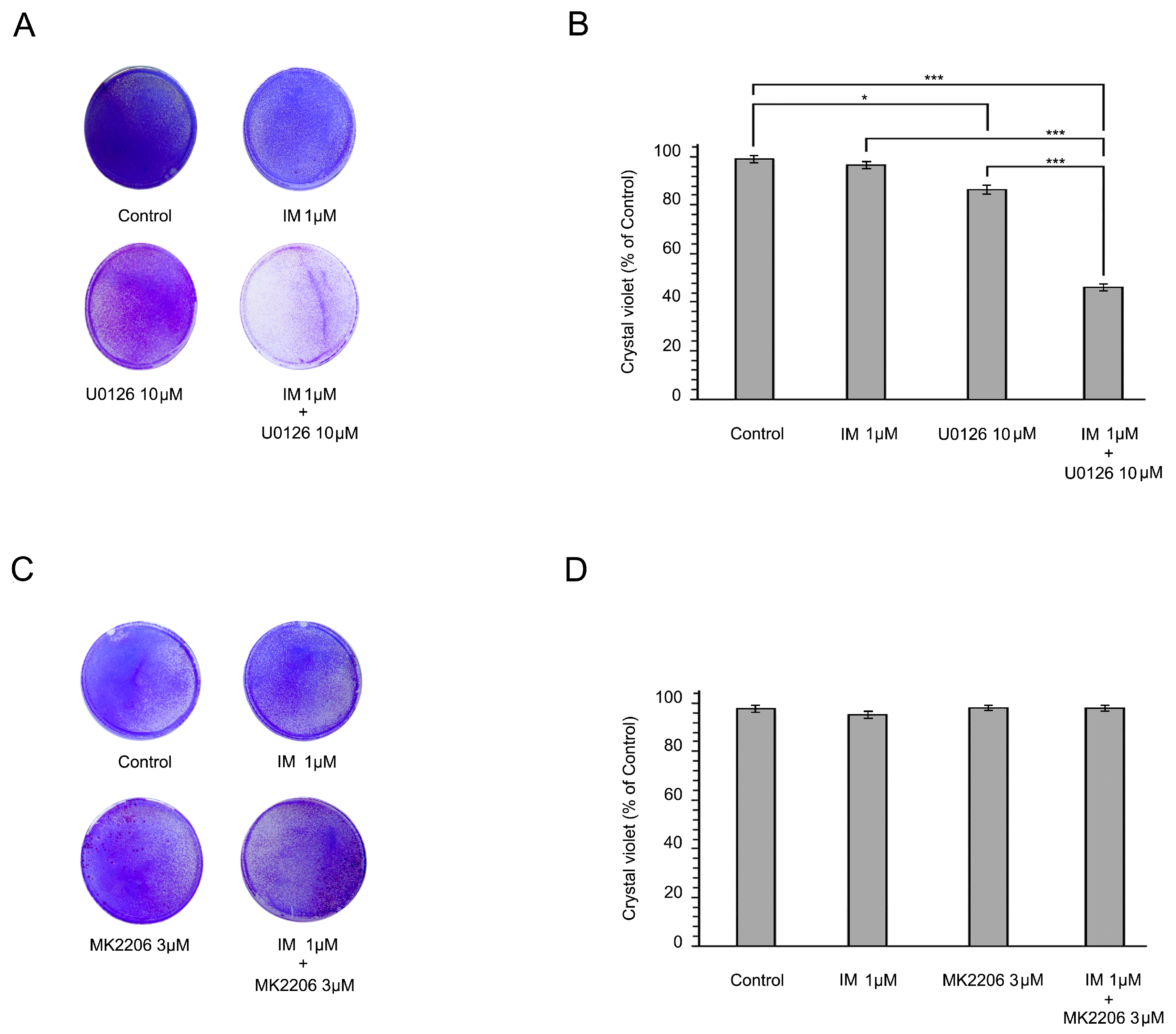
2.3. Inhibition of the RAF/MAPK-Signaling Pathway Restores the Sensitivity of IM-Resistant GIST Cells to IM
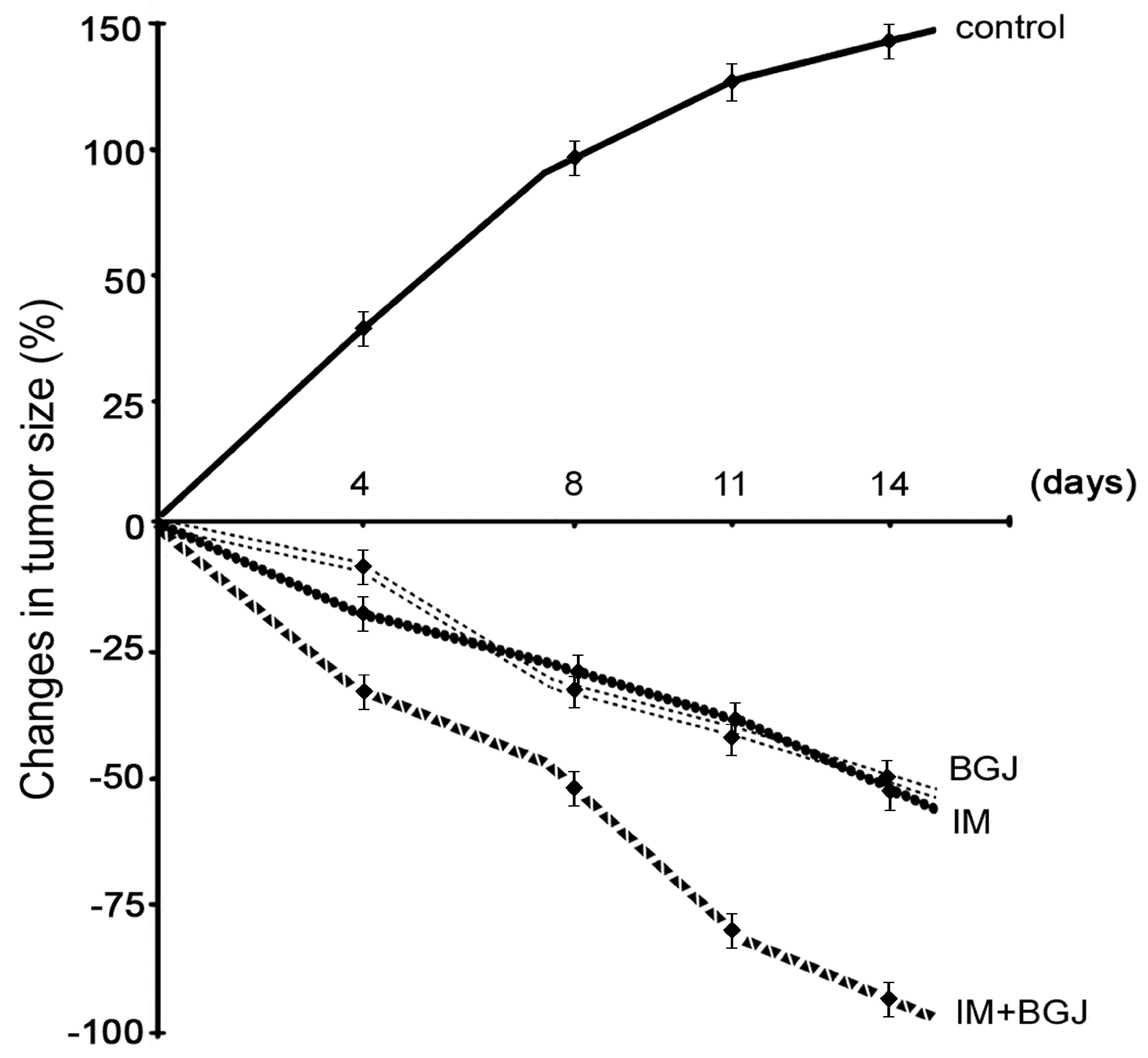
2.4. Inhibition of FGFR Signaling Potentiates IM-Induced Growth Inhibition of GISTs In Vivo
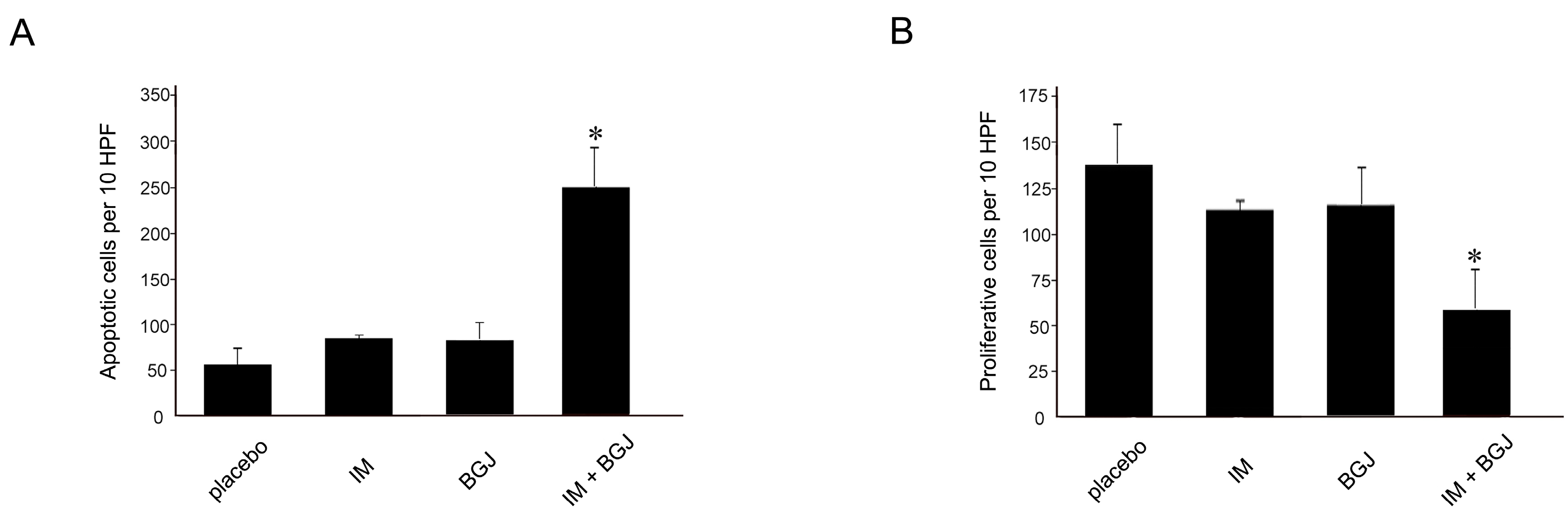
2.5. BGJ398 Enhances the Pro-Apoptotic and Anti-Proliferative Activity of IM in GISTs In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemical Compounds
4.2. Antibodies
4.3. Cell Lines and Culture Conditions
4.4. Phospho-Kinase Array
4.5. Colony Formation Assay
4.6. RNA Extraction and Real-Time Quantitative PCR
4.7. GIST Xenograft Models
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.; Turner, N. Advances and challenges in targeting FGFR signaling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [Green Version]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed] [Green Version]
- Pollock, P.M.; Gartside, M.G.; Dejeza, L.C.; Powell, M.A.; Mallon, M.A.; Davies, H.; Mohammadi, M.; Futreal, P.A.; Stratton, M.R.; Trent, J.M.; et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007, 26, 7158–7162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Mino-Kenudson, M.; Sequist, L.V.; Tammireddy, S.; Morrissey, L.; Christiani, D.C.; Engelman, J.A.; Iafrate, A.J. FGFR1 amplification in squamous cell carcinoma of the lung. J. Thorac. Oncol. 2012, 7, 1775–1780. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Dubrovska, A.; Salamone, R.J.; Walker, J.R.; Grandinetti, K.B.; Bonamy, G.M.; Orth, A.P.; Elliott, J.; Porta, D.G.; Garcia-Echeverria, C.; et al. FGFR2 promotes breast tumorigenicity through maintenance of breast tumor-initiating cells. PLoS ONE 2013, 8, e51671. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br. J. Cancer. 2012, 106, 727–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative splicing of fibroblast growth factor receptor IgIII loops in cancer. J. Nucleic Acids 2012, 2012, 950508. [Google Scholar] [CrossRef] [PubMed]
- Brunello, E.; Brunelli, M.; Bogina, G.; Calio, A.; Manfrin, E.; Nottegar, A.; Vergine, M.; Molino, A.; Bria, E.; Massari, F.; et al. FGFR-1 amplification in metastatic lymph-nodal and haematogenous lobular breast carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Salvesen, H.B.; Chen, T.-H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, G.E.; Kolarova, T.; O’Brien, N.A.; Winterhoff, B.; Yang, G.; Qi, J.; Qi, Z.; Venkatesan, N.; Ayala, R.; Luo, T.; et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol. Cancer Ther. 2013, 12, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-mediated reactivation of MAPK signaling attenuates antitumor effects of imatinib in gastrointestinal stromal tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A novel receptor tyrosine kinase switch promotes gastrointestinal stromal tumor drug resistance. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Dunaev, P.; Galembikova, A.; Mustafin, I.; Valeeva, E. Inhibition of fibroblast growth factor receptor-signaling sensitizes imatinib-resistant gastrointestinal stromal tumors to low doses of topoisomerase II inhibitors. Anticancer Drugs 2018, 29, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients with Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- McDermott, S.C.; Rodriguez-Ramirez, C.; McDermott, S.P.; Wicha, M.S.; Nör, J.E. FGFR signaling regulates resistance of head and neck cancer stem cells to cisplatin. Oncotarget 2018, 9, 25148–25165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by Fibroblast Growth Factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Shoushtari, A.N.; Qin, L.X.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; McFadyen, C.; Sjoberg, A.; Singer, S.; et al. A phase Ib study of BGJ398, a pan-FGFR kinase inhibitor in combination with imatinib in patients with advanced gastrointestinal stromal tumor. Investig. New Drugs 2018. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell. Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Guris, D.L.; Seo, J.H.; Li, L.; Hammond, J.; Talbot, A.; Imamoto, A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev. Cell. 2006, 10, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Suenaga, A.; Hatakeyama, M.; Taiji, M.; Imamoto, A. Structural and functional basis of a role for CRKL in a Fibroblast Growth Factor 8-induced feed-forward loop. Mol. Cell. Biol. 2009, 29, 3076–3087. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.; Klint, P.; Landgren, E.; Claesson-Welsh, L. Fibroblast Growth Factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J. Biol. Chem. 1999, 274, 25726–25734. [Google Scholar] [CrossRef] [PubMed]
- House, S.L.; Branch, K.; Newman, G.; Doetschman, T.; Schultz, J.J. Cardioprotection induced by cardiac-specific overexpression of Fibroblast Growth Factor-2 is mediated by the MAPK cascade. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2167–H2175. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Porter, D.; Scott, A.; Newman, G.; Doetschman, T.; Schultz Jel, J. The cardioprotective effect of the low molecular weight isoform of Fibroblast Growth Factor-2: The role of JNK signaling. J. Mol. Cell. Cardiol. 2007, 42, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Rouse, J.; Zhang, A.; Cariati, S.; Cohen, P.; Comb, M.J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996, 15, 4629–4642. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.; Dawid, I.B. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci. STKE 2004, 2004, pe17. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, S.; Fujiwara, T.; Matsuzaki, S.; Shingaki, K.; Taniguchi, M.; Miyata, S.; Tohyama, M.; Sakai, Y.; Yano, K.; Hosokawa, K.; et al. bFGF regulates PI3-kinase-Rac1-JNK pathway and promotes fibroblast migration in wound healing. PLoS ONE 2010, 5, e12228. [Google Scholar] [CrossRef] [PubMed]
- Floris, G.; Debiec-Rychter, M.; Sciot, R.; Stefan, C.; Fieuws, S.; Machiels, K.; Atadia, P.; Wozniak, A.; Faa, G.; Schoffski, P. High efficacy of panobinostat towards human gastrointestinal stromal tumors in a xenograft mouse model. Clin. Cancer Res. 2009, 15, 4066–4076. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| FGFR1 | AACCTGCCTTATGTCCAGATCT | AGGGGCGAGGTCATCACTGC |
| FGFR2 | GGCTGCCCTACCTCAAGGTTC | AGTCTGGGGAAGCTGTAATCTC |
| FGFR3 | GCACACCCTACGTTACCGTG | GCCTCGTCAGCCTCCACCAG |
| FGF-2 | GCTCTTAGCAGACATTGGAAG | GTGTGTGCTAACCTTACCT |
| GAPDH | GACCACAGTCCATGCCATCA | TCCACCACCCTGTTGCTGTA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boichuk, S.; Galembikova, A.; Dunaev, P.; Micheeva, E.; Valeeva, E.; Novikova, M.; Khromova, N.; Kopnin, P. Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo. Molecules 2018, 23, 2643. https://doi.org/10.3390/molecules23102643
Boichuk S, Galembikova A, Dunaev P, Micheeva E, Valeeva E, Novikova M, Khromova N, Kopnin P. Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo. Molecules. 2018; 23(10):2643. https://doi.org/10.3390/molecules23102643
Chicago/Turabian StyleBoichuk, Sergei, Aigul Galembikova, Pavel Dunaev, Ekaterina Micheeva, Elena Valeeva, Maria Novikova, Natalya Khromova, and Pavel Kopnin. 2018. "Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo" Molecules 23, no. 10: 2643. https://doi.org/10.3390/molecules23102643
APA StyleBoichuk, S., Galembikova, A., Dunaev, P., Micheeva, E., Valeeva, E., Novikova, M., Khromova, N., & Kopnin, P. (2018). Targeting of FGF-Signaling Re-Sensitizes Gastrointestinal Stromal Tumors (GIST) to Imatinib In Vitro and In Vivo. Molecules, 23(10), 2643. https://doi.org/10.3390/molecules23102643