8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia

Abstract
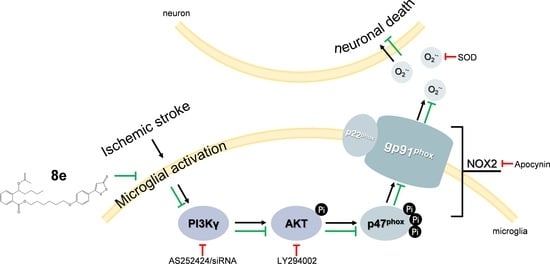
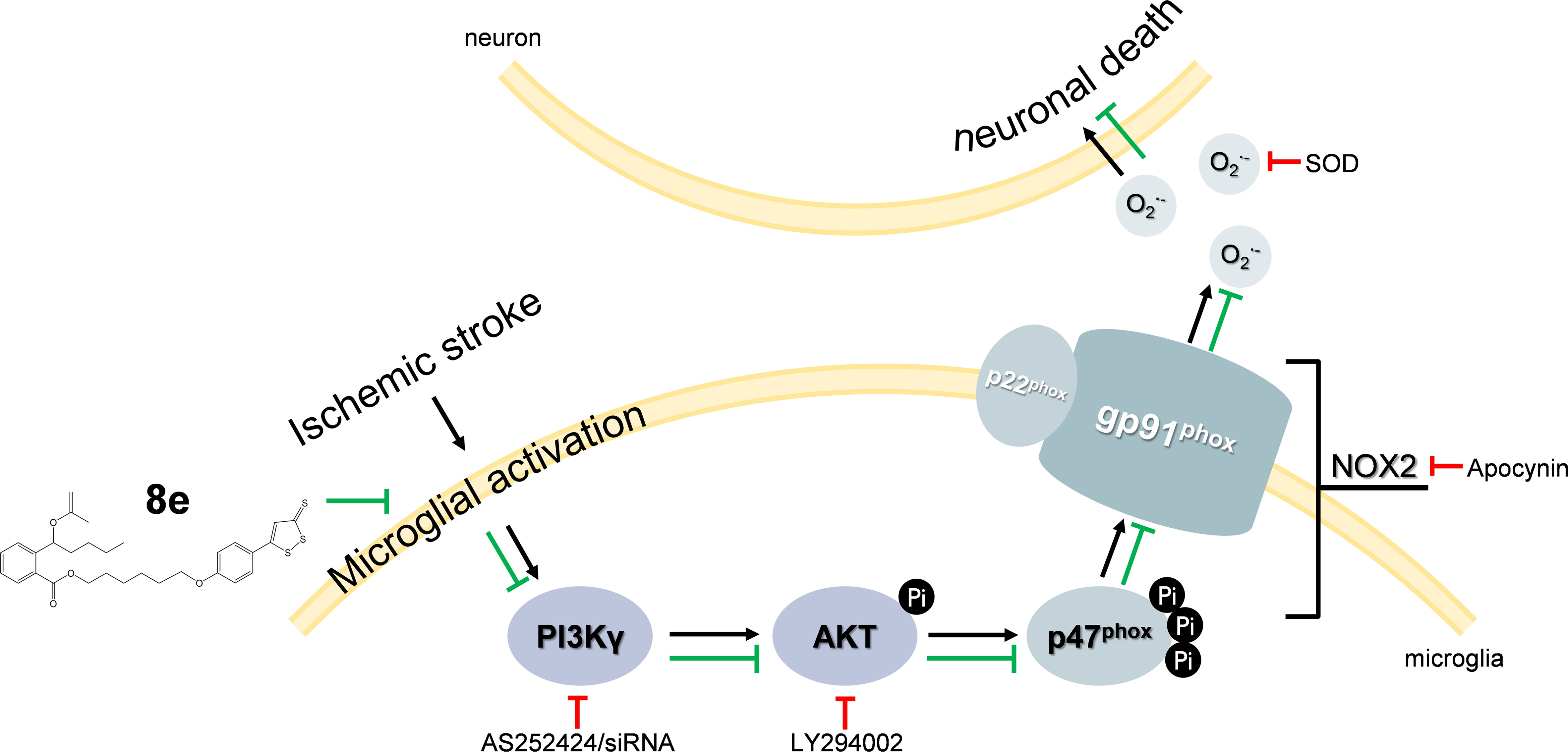
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
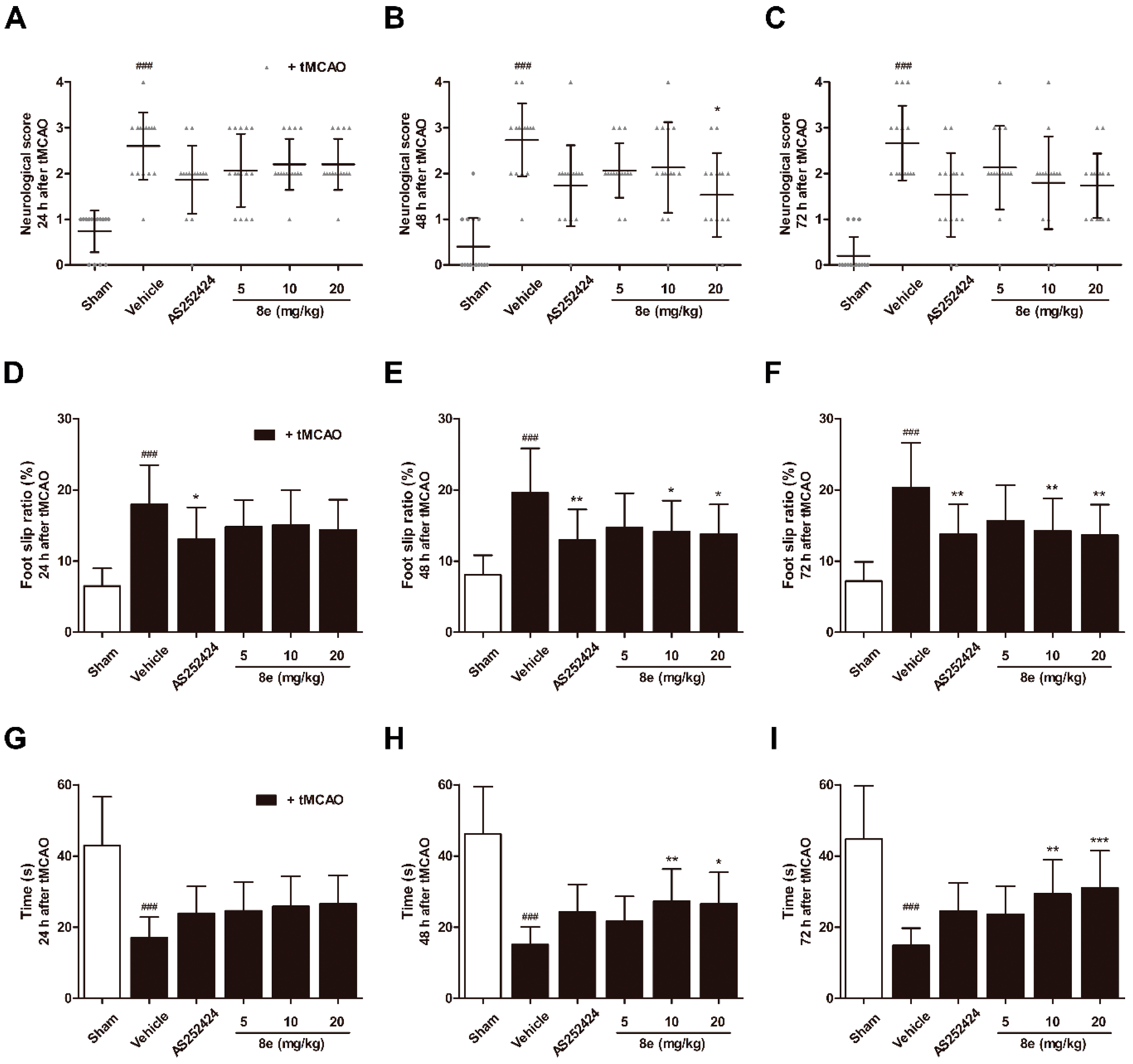
2.1. 8e Enhanced Sensorimotor Function after Cerebral I/R in Rats
2.2. 8e Exerted Protective Effect against Cerebral I/R Injury, Histological Damage and Neural Apoptosis in Rats
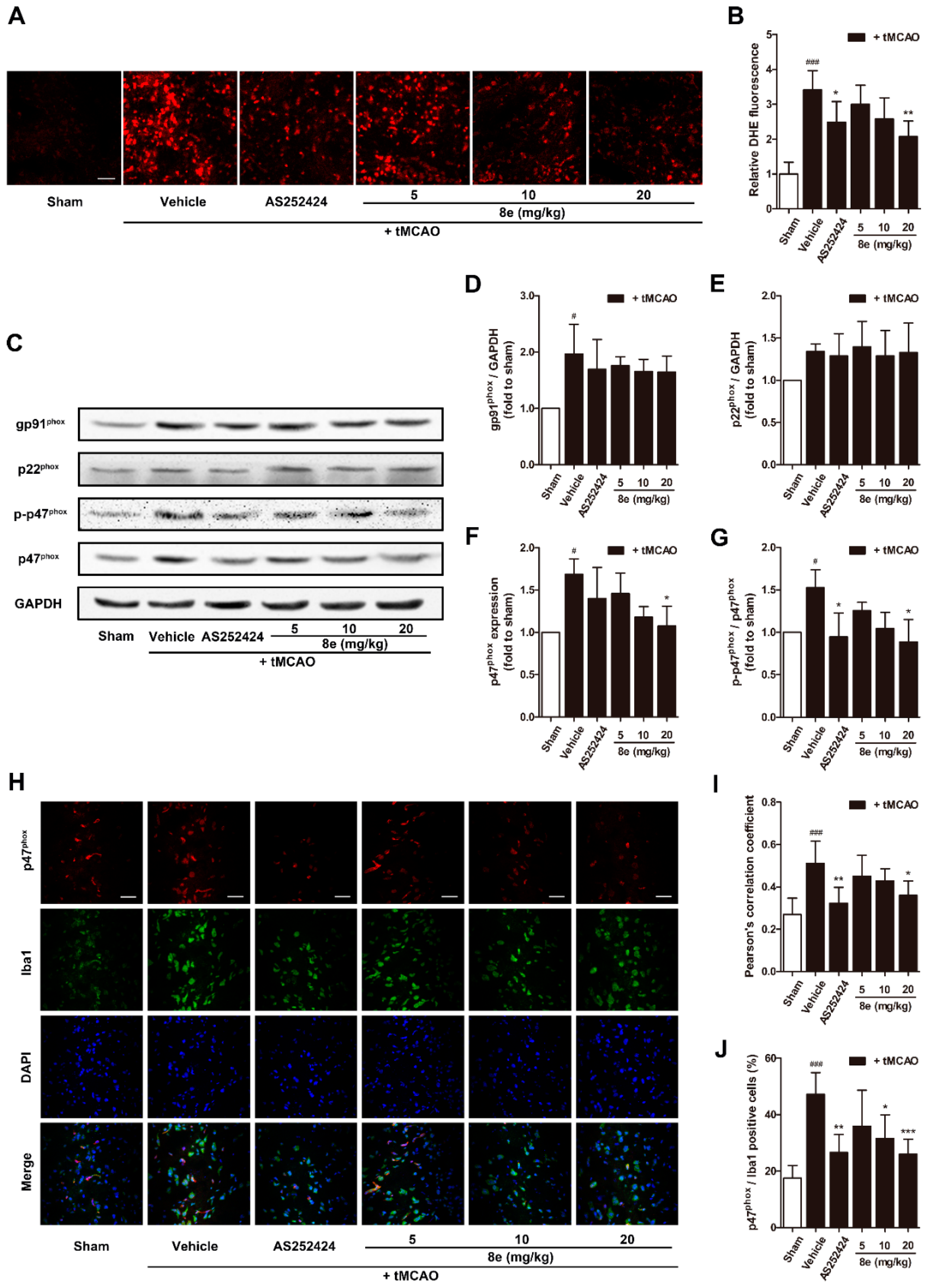
2.3. 8e Attenuated Superoxide Production Following Cerebral I/R in Rats
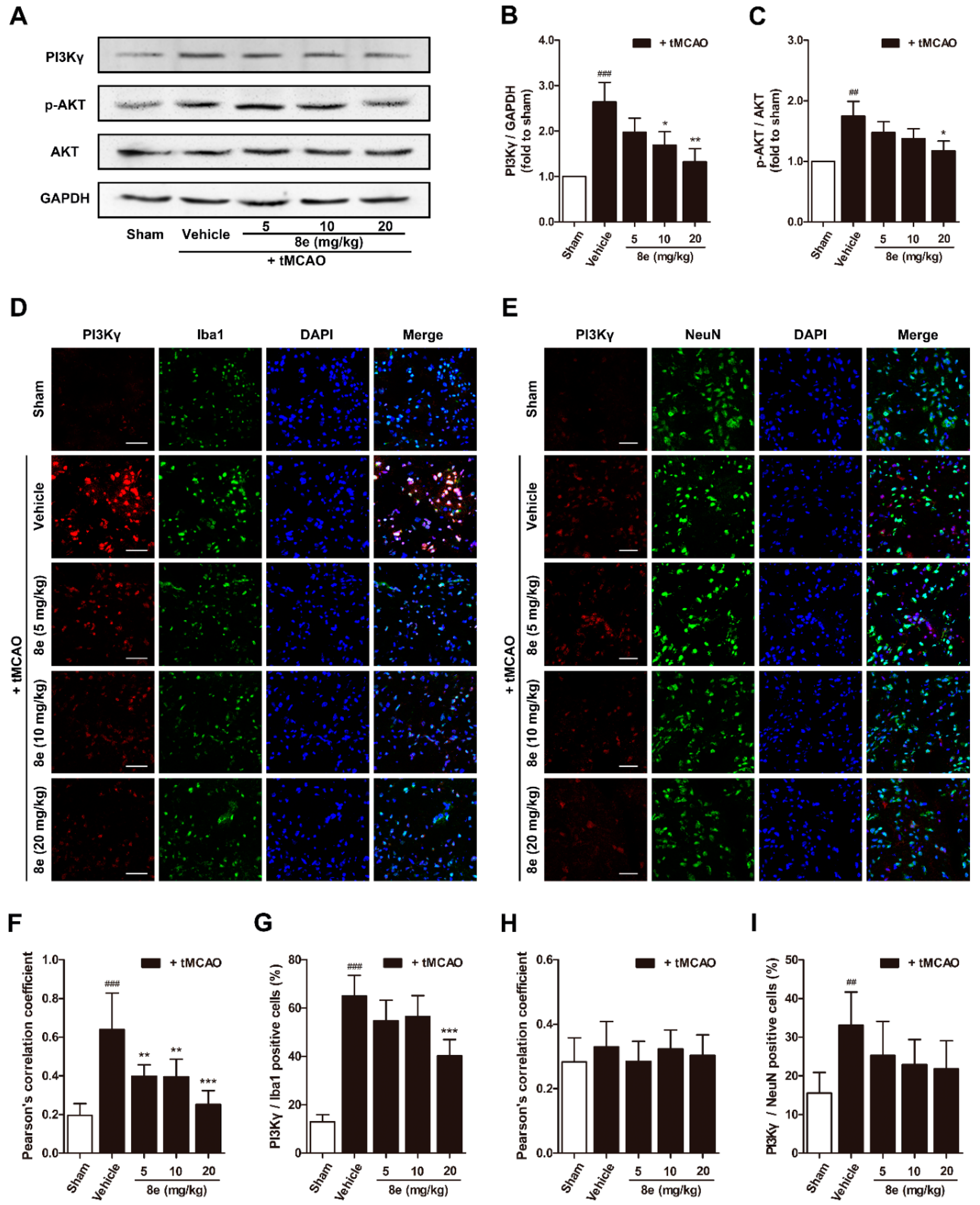
2.4. 8e Inhibited PI3Kγ Signaling in Microglia after Cerebral I/R in Rats
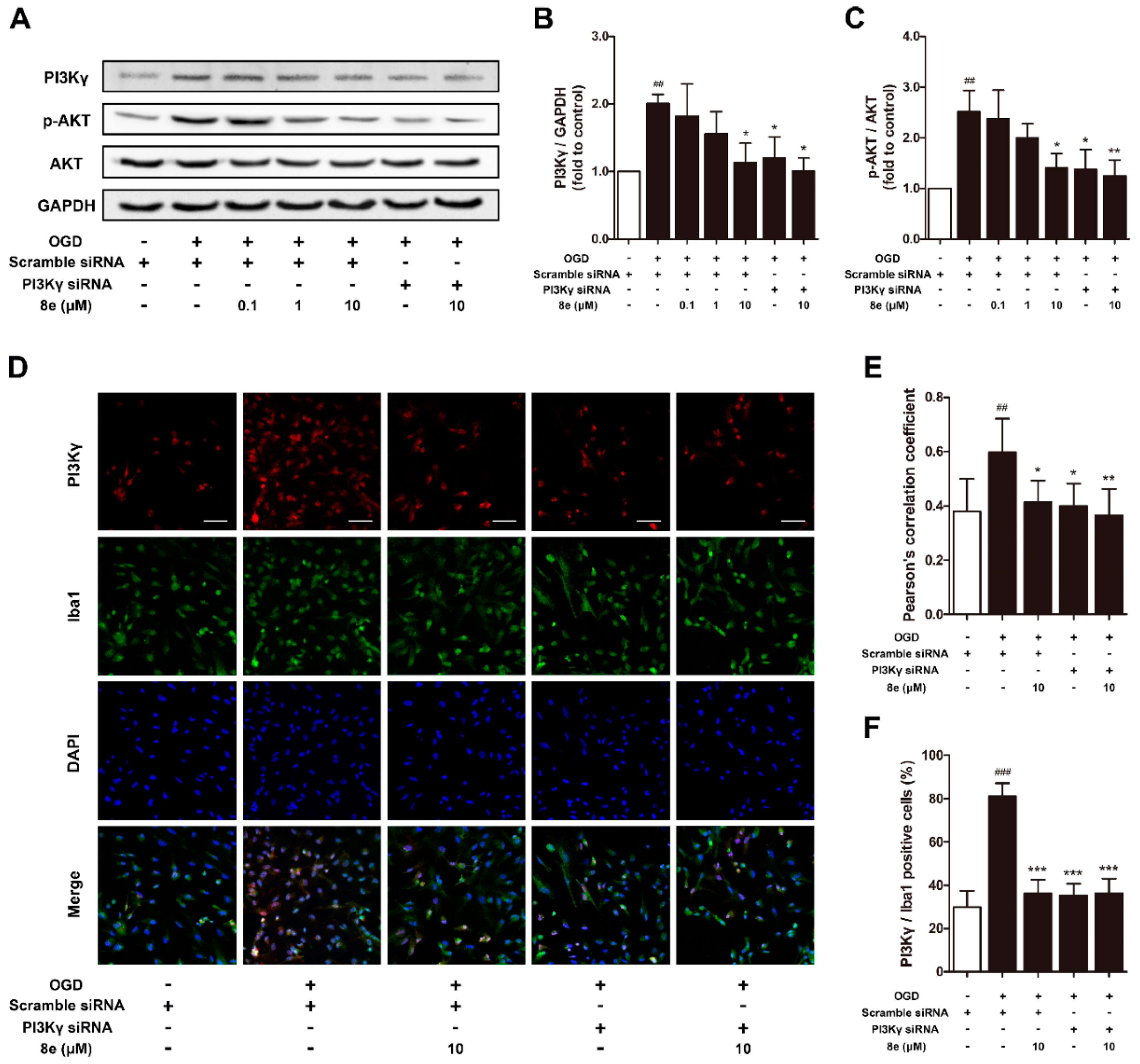
2.5. 8e Down-Regulated PI3Kγ Signaling in OGD-Treated Primary Microglial Cells In Vitro
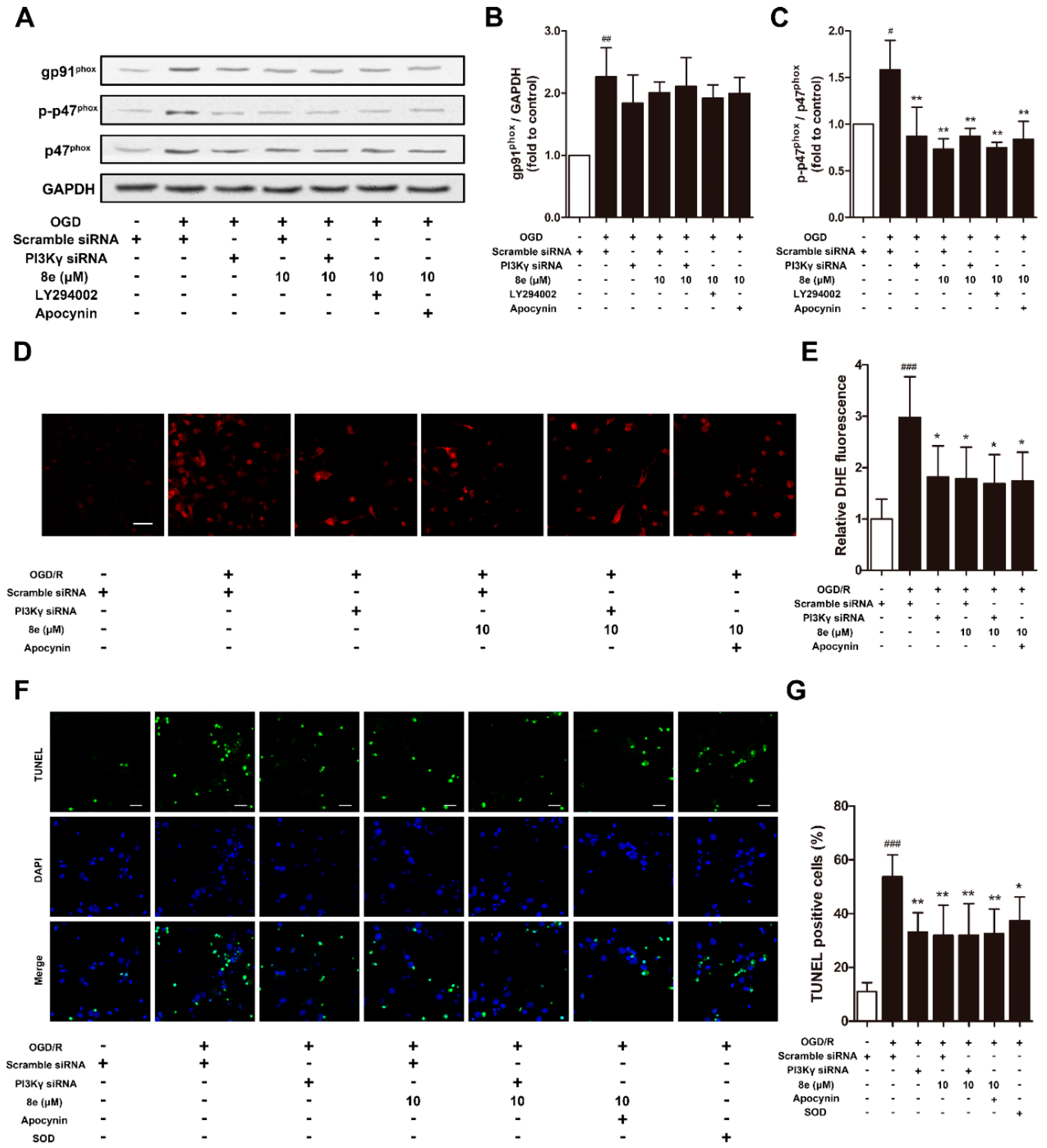
2.6. 8e Modulated NOX2 through PI3Kγ/AKT Signaling in Microglia Subjected to OGD In Vitro
2.7. 8e Protected Neurons against Apoptosis by Down-Regulating Superoxide Production in OGD-Treated Primary Microglia In Vitro
3. Discussion
4. Materials and Methods
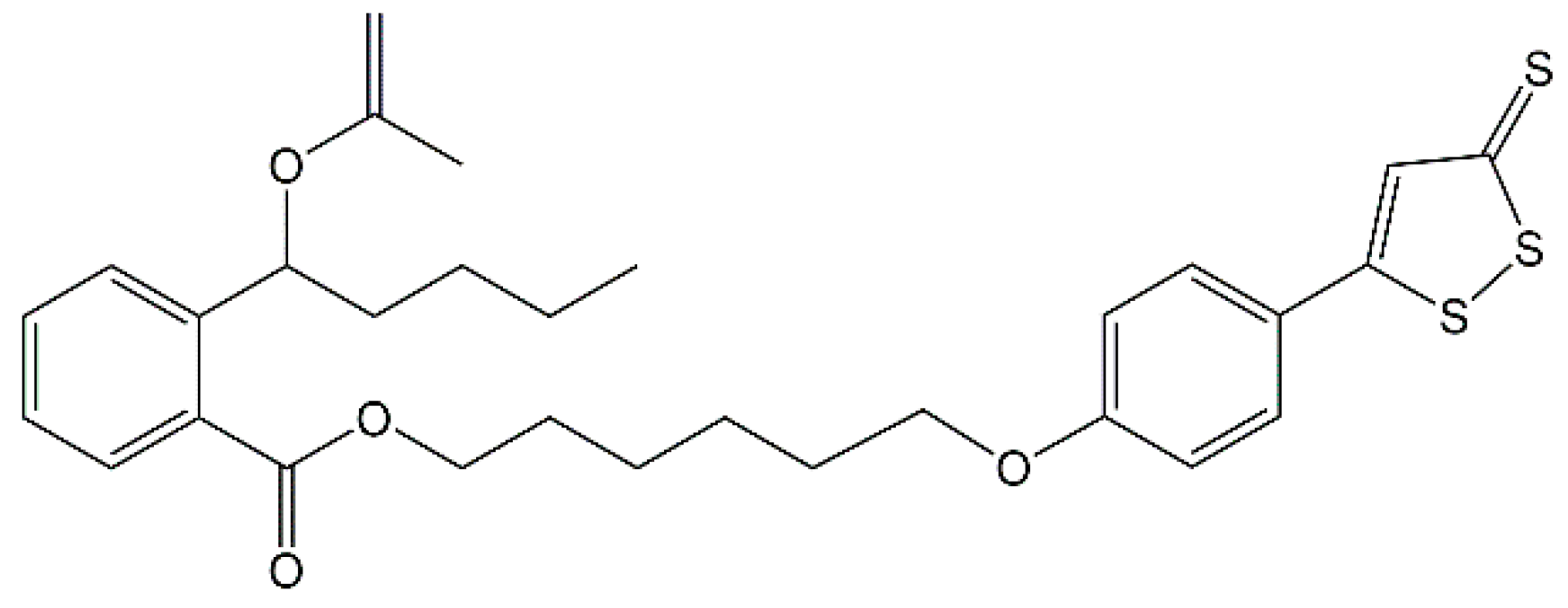
4.1. Chemicals
4.2. Animals
4.3. tMCAO Surgery and Drug Treatment
4.4. Behavioral Assessment
4.4.1. Longa Test
4.4.2. Beam Walking
4.4.3. Prehensile Traction
4.5. Infarct Volume and Brain Edema Measurement
4.6. Primary Microglia Culture
4.7. siRNA Transfection and OGD Treatment
4.8. Primary Neuron Culture and Neuron-Microglia Co-Culture
4.9. Histological Observation and Apoptosis Analysis
4.10. Oxidative Stress Evaluation
4.11. Immunofluorescent Staining
4.12. Western Blotting
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update A Report from the American Heart Association. Circulation 2017, 135, E146–E603. [Google Scholar] [CrossRef] [PubMed]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Davalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Molina, C.A. Reperfusion Therapies for Acute Ischemic Stroke Current Pharmacological and Mechanical Approaches. Stroke 2011, 42, S16–S19. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H. Experimental models, neurovascular mechanisms and translational issues in stroke research. Br. J. Pharmacol. 2008, 153, S396–S405. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. JPPP 2013, 5, 73–90. [Google Scholar]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Butovsky, O.; Bruck, W.; Hanisch, U.K. Microglial phenotype: Is the commitment reversible? Trends Neurosci. 2006, 29, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Biber, K.; Owens, T.; Boddeke, E. What is microglia neurotoxicity (Not)? Glia 2014, 62, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Hoyal, C.R.; Gutierrez, A.; Young, B.M.; Catz, S.D.; Lin, J.H.; Tsichlis, P.N.; Babior, B.M. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 5130–5135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Browning, E.A.; Hong, N.K.; DeBolt, K.; Sorokina, E.M.; Liu, W.D.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart C 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okkenhaug, K. Signaling by the Phosphoinositide 3-Kinase Family in Immune Cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, M.; Ruckle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Francon, B.; et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.F.; Bartolome, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Ruckle, T.; Schwarz, M.K.; Rodriguez, S.; et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Katare, R.; Meloni, M.; Damilano, F.; Hirsch, E.; Emanueli, C.; Madeddu, P. Involvement of Phosphoinositide 3-Kinase gamma in Angiogenesis and Healing of Experimental Myocardial Infarction in Mice. Circ. Res. 2010, 106, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ruckle, T.; Schwarz, M.K.; Rommel, C. PI3K gamma inhibition: Towards an ‘aspirin of the 21st century’? Nat. Rev. Drug Discov. 2006, 5, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Passos, G.F.; Figueiredo, C.P.; Prediger, R.D.S.; Silva, K.A.B.S.; Siqueira, J.M.; Duarte, F.S.; Leal, P.C.; Medeiros, R.; Calixto, J.B. Involvement of phosphoinositide 3-kinase gamma in the neuro-inflammatory response and cognitive impairments induced by beta-amyloid 1–40 peptide in mice. Brain Behav. Immun. 2010, 24, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Sherchan, P.; Wang, Y.; Reis, C.; Applegate, R.L., 2nd; Tang, J.; Zhang, J.H. Phosphoinositide 3-Kinase Gamma Contributes to Neuroinflammation in a Rat Model of Surgical Brain Injury. J. Neurosci. 2015, 35, 10390–10401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Yu, S.; Song, Z.; Quillin, J.W.; Deasis, D.P.; Penninger, J.M.; Nanda, A.; Granger, D.N.; Li, G. Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem. Biophys. Res. Commun. 2010, 399, 458–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Song, Z.F.; Yu, S.Y.; Piazza, A.; Nanda, A.; Penninger, J.M.; Granger, D.N.; Li, G.H. Phosphatidylinositol-3-Kinase Gamma Plays a Central Role in Blood-Brain Barrier Dysfunction in Acute Experimental Stroke. Stroke 2011, 42, 2033–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Frahm, C.; Schneble, N.; Muller, J.P.; Brodhun, M.; Franco, I.; Witte, O.W.; Hirsch, E.; Wetzker, R.; Bauer, R. Phosphoinositide 3-Kinase gamma Restrains Neurotoxic Effects of Microglia After Focal Brain Ischemia. Mol. Neurobiol. 2016, 53, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Lee, H.R.; Sim, S.E.; Baek, J.; Yu, N.K.; Choi, J.H.; Ko, H.G.; Lee, Y.S.; Park, S.W.; Kwak, C.; et al. PI3K gamma is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat. Neurosci. 2011, 14, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen Sulfide and Cell Signaling. Annu. Rev. Pharmacol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yao, X.; Zhang, Y.; Li, W.; Kang, K.; Sun, L.; Sun, X. The protective role of hydrogen sulfide in myocardial ischemia-reperfusion-induced injury in diabetic rats. Int. J. Cardiol. 2011, 152, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.P.; Hosgood, S.A.; Patel, M.; Rose, R.; Read, K.; Nicholson, M.L. Effects of hydrogen sulphide in an experimental model of renal ischaemia-reperfusion injury. Br. J. Surg. 2012, 99, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Marutani, E.; Kosugi, S.; Tokuda, K.; Khatri, A.; Nguyen, R.; Atochin, D.N.; Kida, K.; Van Leyen, K.; Arai, K.; Ichinose, F. A novel hydrogen sulfide-releasing N-methyl-D-aspartate receptor antagonist prevents ischemic neuronal death. J. Biol. Chem. 2012, 287, 32124–32135. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Sheng, X.; Huang, Z.; Li, T.; Zhang, M.; Xu, J.; Ji, H.; Yin, J.; Zhang, Y. Design, synthesis and biological evaluation of hydrogen sulfide releasing derivatives of 3-n-butylphthalide as potential antiplatelet and antithrombotic agents. Org. Biomol. Chem. 2014, 12, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cao, Y.; Ao, G.; Hu, L.; Liu, H.; Wu, J.; Wang, X.; Jin, M.; Zheng, S.; Zhen, X.; et al. CaMKKbeta-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid. Redox Signal. 2014, 21, 1741–1758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Jia, J.; Ao, G.Z.; Hu, L.F.; Liu, H.; Xiao, Y.Q.; Du, H.P.; Alkayed, N.J.; Liu, C.F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; McGeer, E.; Kodela, R.; Kashfi, K.; McGeer, P.L. NOSH-aspirin (NBS-1120), a novel nitric oxide and hydrogen sulfide releasing hybrid, attenuates neuroinflammation induced by microglial and astrocytic activation: A new candidate for treatment of neurodegenerative disorders. Glia 2013, 61, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yuan, L.; Liu, D.X.; Wang, J.M.; Wang, S.L.; Zhang, Q.R.; Gong, Y.F.; Liu, H.D.; Hao, A.J.; Wang, Z. Hydrogen sulfide attenuates hypoxia-induced neurotoxicity through inhibiting microglial activation. Pharmacol. Res. 2014, 84, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Sodium thiosulfate attenuates glial-mediated neuroinflammation in degenerative neurological diseases. J. Neuroinflamm. 2016, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.M.; Chen, G.W.; Liu, Y.; Xiao, X.Z. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Lu, M.; Tiong, C.X.; Dawe, G.S.; Hu, G.; Bian, J.S. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 2010, 9, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Z.; Shi, M.M.; Xie, L.; Wu, Z.Y.; Li, G.; Hua, F.; Bian, J.S. Sulfhydration of p66Shc at Cysteine59 Mediates the Antioxidant Effect of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 21, 2531–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.L.; Huang, X.W.; Wang, Y.G.; Cao, Y.X.; Zhang, C.C.; Zhu, Y.C. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3 beta-dependent opening of mPTP. Am. J. Physiol. Heart C 2010, 298, H1310–H1319. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Biggs, T.D.; Xian, M. Hydrogen sulfide (H2S) releasing agents: Chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.H.; Parkinson, D.B.; Moore, P.K. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, K.; Chen, C.P.L.H.; Halliwell, B.; Moore, P.K.; Wong, P.T.H. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006, 37, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Olson, K.R. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 2013, 85, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Tazzari, V.; Giustarini, D.; Rossi, R.; Sparatore, A.; Del Soldato, P.; McGeer, E.; McGeer, P.L. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: Potential for treating Parkinson disease. J. Biol. Chem. 2010, 285, 17318–17328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Sparatore, A.; Del Soldato, P.; Bian, J.S. ACS84, a novel hydrogen sulfide-releasing compound, protects against amyloid beta-induced cell cytotoxicity. Neurochem. Int. 2011, 58, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Bachour, S.P.; Hevesi, M.; Bachour, O.; Sweis, B.M.; Mahmoudi, J.; Brekke, J.A.; Divani, A.A. Comparisons between Garcia, Modo, and Longa rodent stroke scales: Optimizing resource allocation in rat models of focal middle cerebral artery occlusion. J. Neurol. Sci. 2016, 364, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zausinger, S.; Hungerhuber, E.; Baethmann, A.; Reulen, H.J.; Schmid-Elsaesser, R. Neurological impairment in rats after transient middle cerebral artery occlusion: A comparative study under various treatment paradigms. Brain Res. 2000, 863, 94–105. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Koerner, I.P.; Moller, T. Microglia in ischemic brain injury. Future Neurol. 2010, 5, 227–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Yang, G.J.; Li, G.H. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukocyte Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Strecker, J.K.; Schabitz, W.R.; Ringelstein, E.B.; Kiefer, R. Effects of monocyte chemoattractant protein 1 on blood-borne cell recruitment after transient focal cerebral ischemia in mice. Neuroscience 2009, 161, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Besselmann, M.; Leonhard, C.; Mueller, M.; Ringelstein, E.B.; Kiefer, R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2003, 183, 25–33. [Google Scholar] [CrossRef]
- Schilling, M.; Besselmann, M.; Muller, M.; Strecker, J.K.; Ringelstein, E.B.; Kiefer, R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2005, 196, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Weston, R.M.; Jones, N.M.; Jarrott, B.; Callaway, J.K. Inflammatory cell infiltration after endothelin-1-induced cerebral ischemia: Histochemical and myeloperoxidase correlation with temporal changes in brain injury. J. Cereb. Blood Flow Metab. 2007, 27, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Li, M.L.; Tian, W.K.; Wang, S.W.; Cui, L.Z.; Li, H.; Wang, H.J.; Ji, A.L.; Li, Y.Z. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci. Rep. 2017, 7, 5134. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Brandes, R.P. Which NADPH Oxidase Isoform Is Relevant for Ischemic Stroke? The Case for Nox 2. Antioxid. Redox Signal. 2013, 18, 1400–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, F.; Teixeira, P.C.; Braunersreuther, V.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiology and Treatments of Oxidative Injury in Ischemic Stroke: Focus on the Phagocytic NADPH Oxidase 2. Antioxid. Redox Signal. 2015, 23, 460–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.P.; Cairns, B.; Rae, J.; Errett-Baroncini, C.; Hongo, J.A.S.; Erickson, R.W.; Curnutte, J.T. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 2001, 21, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Wang, L.N.; Li, T.T.; Huang, Z.J.; Lai, Y.S.; Ji, H.; Wan, X.L.; Xu, J.Y.; Tian, J.D.; Zhang, Y.H. Novel Hybrids of Optically Active Ring-Opened 3-n-Butylphthalide Derivative and Isosorbide as Potential Anti-Ischemic Stroke Agents. J. Med. Chem. 2013, 56, 3078–3089. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Wang, J.; Fang, Y.Q.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.F.; Liao, H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Biernaskie, J.; Chernenko, G.; Corbett, D. Efficacy of rehabilitative experience declines with time after focal ischemic brain injury. J. Neurosci. 2004, 24, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Huang, P.L.; Panahian, N.; Fishman, M.C.; Moskowitz, M.A. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J. Cereb. Blood Flow Metab. 1996, 16, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Schlichter, L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008, 28, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Kim, S.J.; Song, E.C.; Kim, E.H.; Park, D.K.; Sinn, D.I.; Kim, J.M.; Kim, M.; et al. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral Hemorrhage. J. Pharmacol. Exp. Ther. 2007, 322, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, T.; Yu, H.; Shen, H.; Xia, W. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J. Neuroinflamm. 2014, 11, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Xiang, P.; Li, T.; Lan, L.; Xu, X.; Lu, G.; Ji, H.; Zhang, Y.; Li, Y. NOSH-NBP, a Novel Nitric Oxide and Hydrogen Sulfide- Releasing Hybrid, Attenuates Ischemic Stroke-Induced Neuroinflammatory Injury by Modulating Microglia Polarization. Front. Cell. Neurosci. 2017, 11, 154. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound 8e is not available from the authors. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, X.; Li, T.; Zhang, Y.; Ji, H. 8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia. Molecules 2018, 23, 2828. https://doi.org/10.3390/molecules23112828
Wang L, Wang X, Li T, Zhang Y, Ji H. 8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia. Molecules. 2018; 23(11):2828. https://doi.org/10.3390/molecules23112828
Chicago/Turabian StyleWang, Linna, Xiaoli Wang, Tingting Li, Yihua Zhang, and Hui Ji. 2018. "8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia" Molecules 23, no. 11: 2828. https://doi.org/10.3390/molecules23112828
APA StyleWang, L., Wang, X., Li, T., Zhang, Y., & Ji, H. (2018). 8e Protects against Acute Cerebral Ischemia by Inhibition of PI3Kγ-Mediated Superoxide Generation in Microglia. Molecules, 23(11), 2828. https://doi.org/10.3390/molecules23112828