Interfacing CRYSTAL/AMBER to Optimize QM/MM Lennard–Jones Parameters for Water and to Study Solvation of TiO2 Nanoparticles

 , and
, and
Abstract
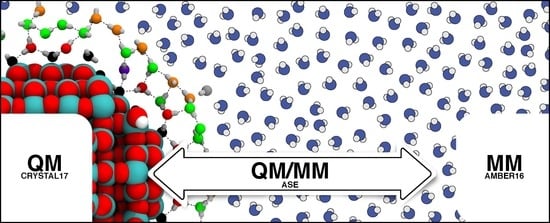
:
1. Introduction
2. Results
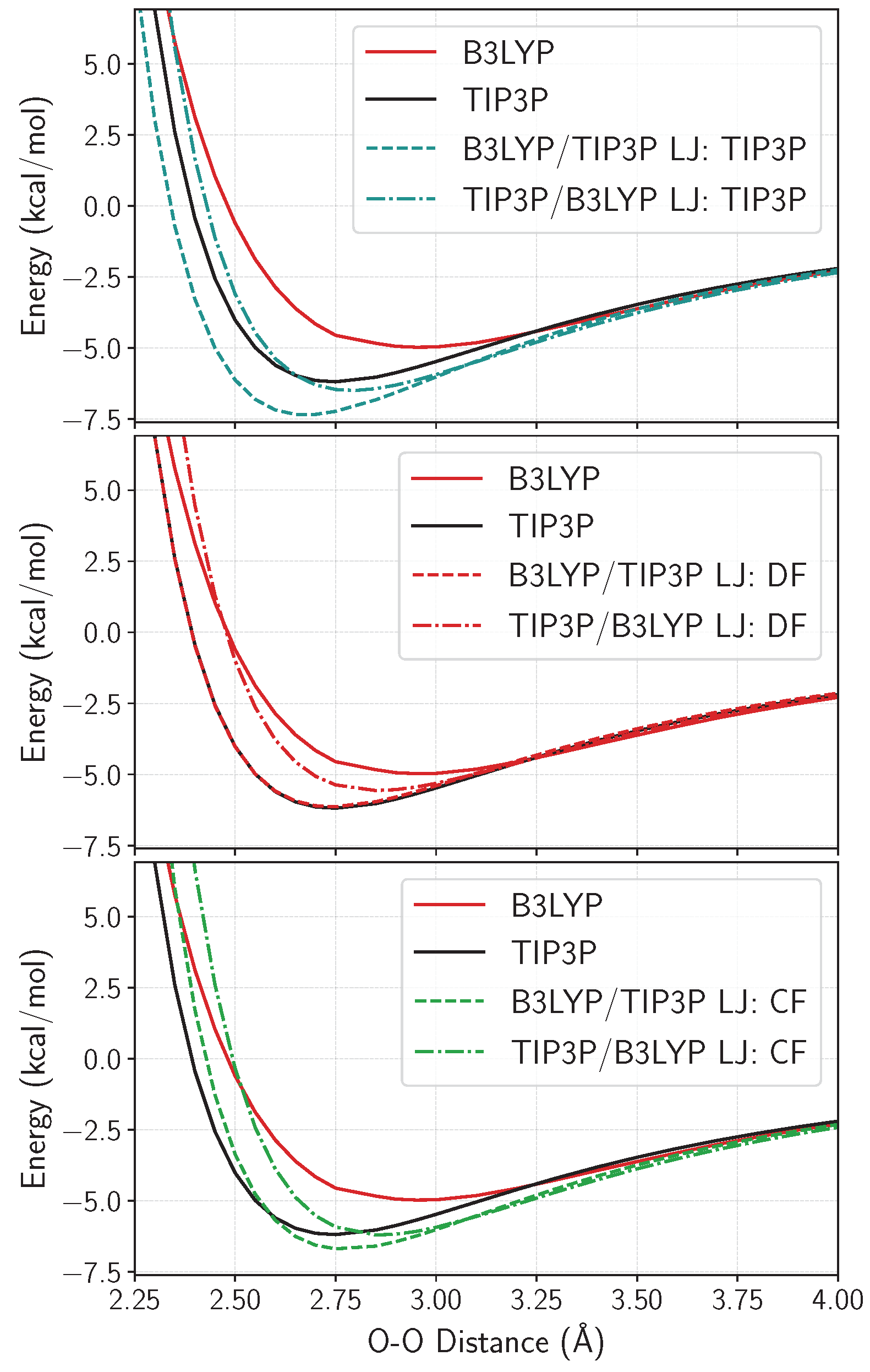
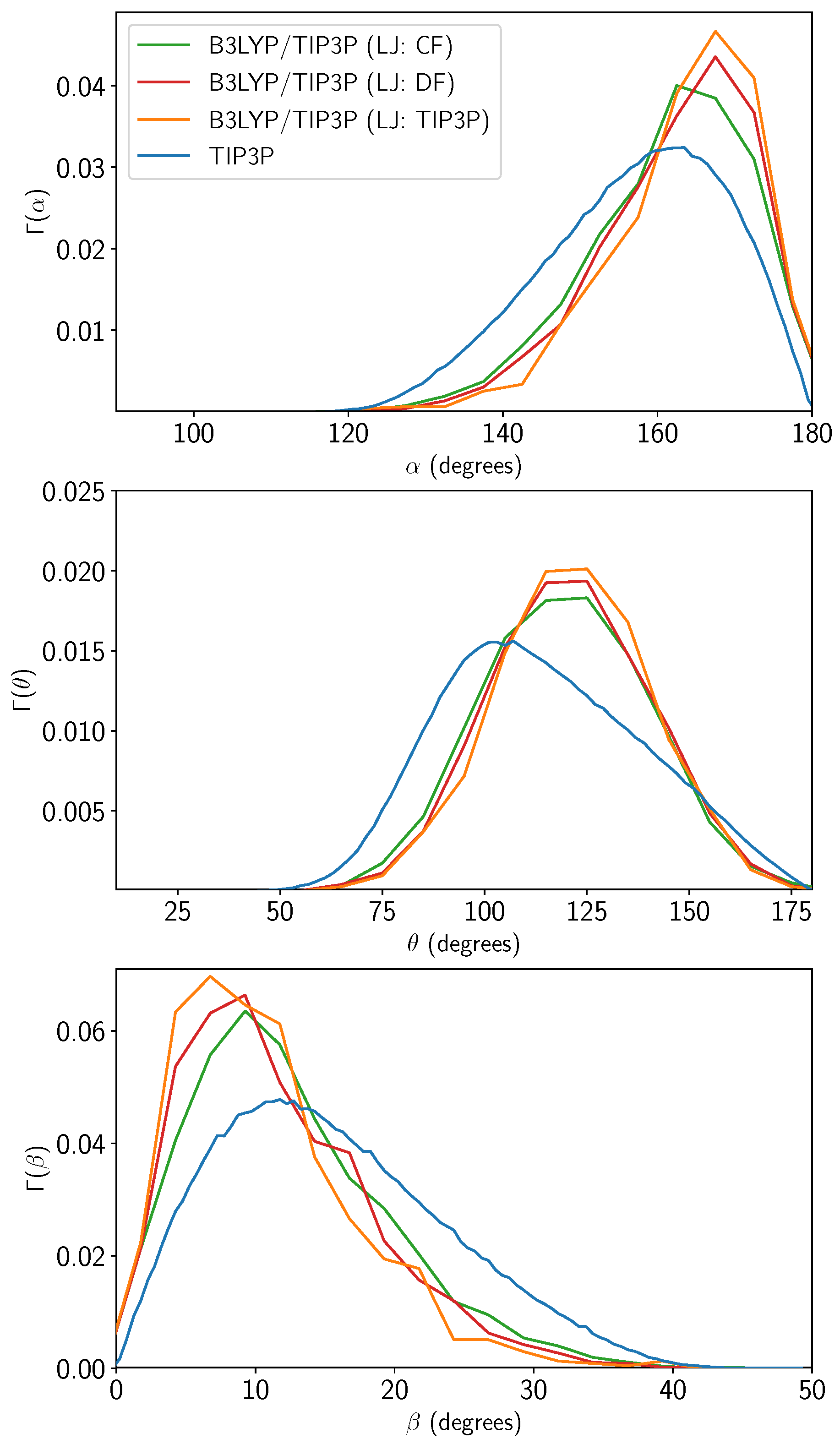
2.1. Optimizing the Lennard–Jones Parameters for QM/MM Water–Water Interactions
2.1.1. Water Dimer
2.1.2. Water Clusters
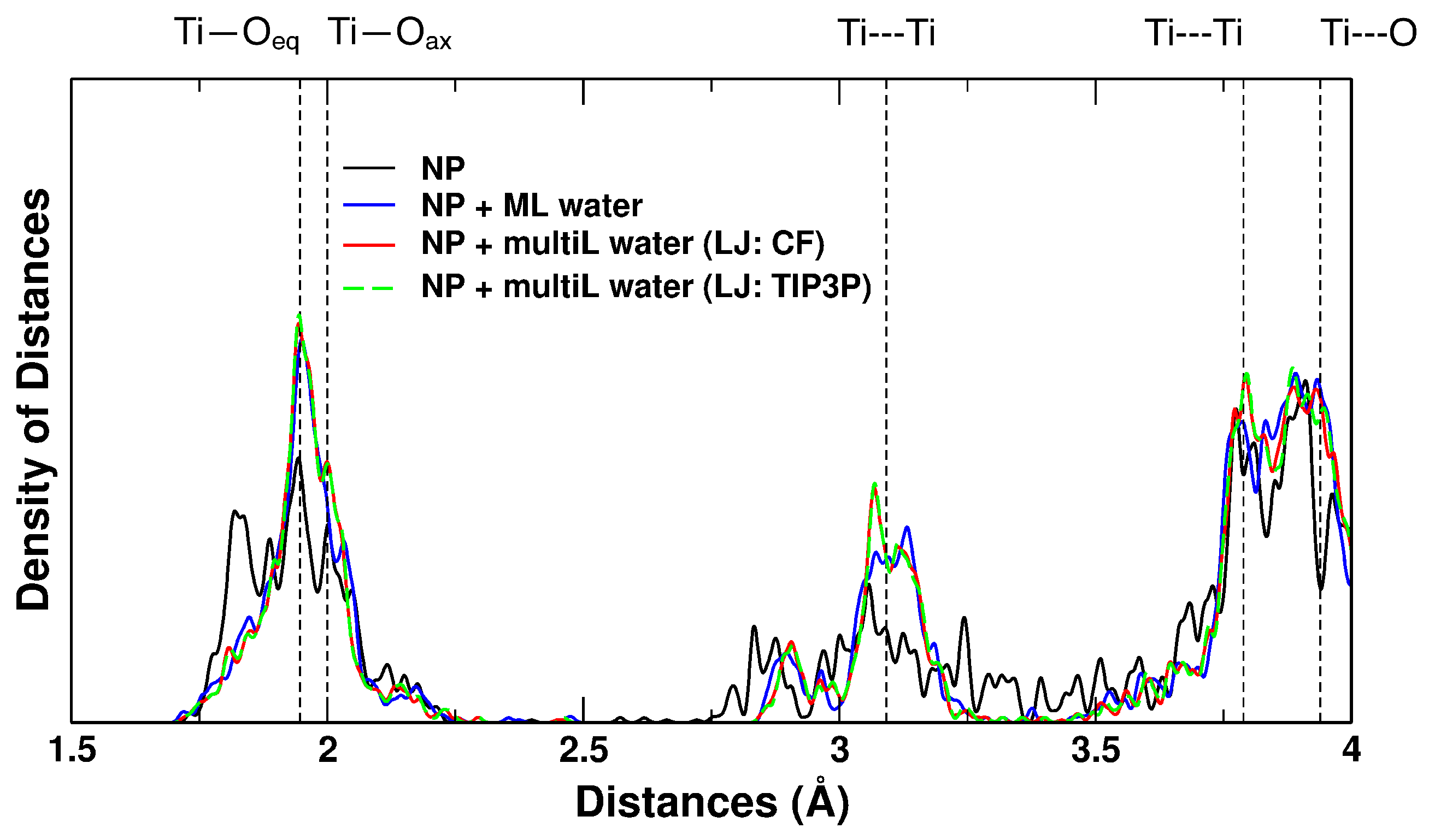
2.2. Nanoparticles
3. Discussion
3.1. Water–Water QM/MM LJ Re-Parameterization and Parameter Transferability Tests
3.2. Nanoparticles
4. Materials and Methods
4.1. The Basics of Electrostatic Embedding QM/MM
4.2. Computational Details
4.2.1. Water Dimers and Clusters
4.2.2. Fitting Strategies
4.2.3. Liquid Water Simulations
4.2.4. Organic Molecule/Water Dimer Benchmarks
4.2.5. Nanoparticle Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADF | Angular Distribution Function |
| AIMD | Ab Initio Molecular Dynamics |
| BOMD | Born–Oppenheimer Molecular Dynamics |
| CF | Cluster Fit |
| DF | Dimer Fit |
| DOS | Density of States |
| GGA | Generalized Gradient Approximation |
| HF | Hartree-Fock |
| IQR | Interquartile Range |
| LCAO | Linear Combination of Atomic Orbitals |
| LJ | Lennard–Jones |
| ML | Monolayer |
| multiL | Multilayer |
| NP | Nanoparticle |
| PDOS | Projected Density of States |
| QM/MM | Quantum Mechanical/Molecular Mechanical |
| RDF | Radial Distribution Function |
| RMSD | Root Mean Square Deviation |
| vdW | van der Waals |
Appendix A. Further Water QM/MM Benchmarks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | rOO (Å) | ∠(α) (deg.) | ∠(OOH) (deg.) | ΔEint (kcal/mol) |
|---|---|---|---|---|
| B3LYP | 2.8803 | 51.7 | 6.3 | −5.08 |
| B3LYP/TIP3P (LJ: TIP3P) | 2.6414 | 15.9 | 0.4 | −8.76 |
| B3LYP/TIP3P (LJ: DF) | 2.7039 | 21.6 | 2.0 | −7.31 |
| B3LYP/TIP3P (LJ: CF) | 2.7414 | 17.8 | 0.4 | −7.84 |
| TIP3P/B3LYP (LJ: TIP3P) | 2.8578 | 61.3 | 0.1 | −6.51 |
| TIP3P/B3LYP (LJ: DF) | 2.8502 | 72.2 | 3.2 | −5.49 |
| TIP3P/B3LYP (LJ: CF) | 2.7859 | 61.0 | 0.1 | −6.22 |
| TIP3P | 2.7461 | 20.3 | 4.4 | −6.54 |
| CCSD(T)/cc-pVQZ a | 2.9104 | 60.3 | 4.8 | −5.02 |
Appendix B. Organic Molecule/Water Dimer Benchmark
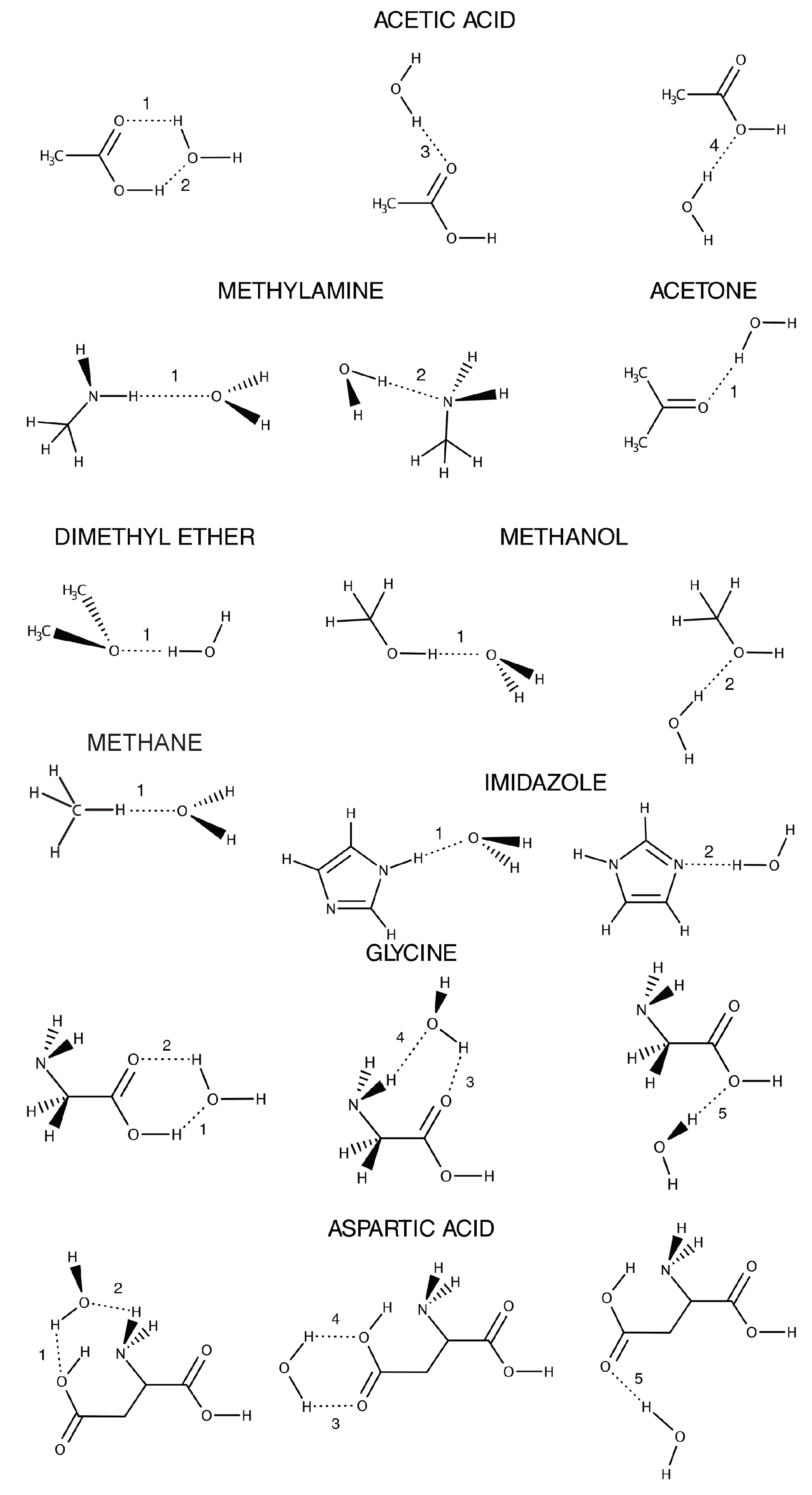
Appendix B.1. Structures and Nomenclature

Appendix B.2. Benchmark Results
| Molecule | HB | B3LYP | B3LYP/TIP3P | ||||
|---|---|---|---|---|---|---|---|
| EHB | rHB | φHB | EHB | rHB | φHB | ||
| Acetic Acid | 1 | 10.50 | 1.782 | 156.9 | 9.34 | 1.972 | 160.8 |
| 2 | 10.50 | 1.979 | 136.0 | 9.34 | 2.198 | 135.0 | |
| 3 | 5.94 | 1.930 | 160.0 | 6.03 | 1.987 | 160.2 | |
| 4 | 3.58 | 2.067 | 153.6 | 3.97 | 2.081 | 153.7 | |
| Methyl Amine | 1 | 2.68 | 2.188 | 158.7 | 3.20 | 2.175 | 166.3 |
| 2 | 7.90 | 1.908 | 171.2 | 6.59 | 2.055 | 173.4 | |
| Acetone | 1 | 6.31 | 1.907 | 164.6 | 6.47 | 1.948 | 164.9 |
| Dimethyl Ether | 1 | 5.24 | 1.907 | 175.6 | 5.05 | 1.952 | 166.1 |
| Methanol | 1 | 5.58 | 1.936 | 173.9 | 5.93 | 1.946 | 172.4 |
| 2 | 5.96 | 1.898 | 177.3 | 6.04 | 1.932 | 176.8 | |
| Imidazole | 1 | 6.50 | 1.953 | 179.2 | 6.77 | 1.964 | 178.5 |
| 2 | 7.08 | 1.949 | 179.4 | 6.05 | 2.092 | 177.0 | |
| Methane | 1 | 0.38 | 2.566 | 156.7 | 0.74 | 2.660 | 163.6 |
| Glycine | 1 | 10.50 | 1.776 | 156.6 | 9.29 | 1.959 | 160.3 |
| 2 | 10.50 | 1.995 | 135.0 | 9.29 | 2.204 | 134.4 | |
| 3 | 6.34 | 1.943 | 156.8 | 7.05 | 2.019 | 156.7 | |
| 4 | 6.34 | 2.186 | 146.1 | 7.05 | 2.167 | 156.0 | |
| 5 | 3.26 | 2.045 | 152.2 | 3.80 | 2.101 | 152.9 | |
| Aspartic acid | 1 | 6.56 | 2.155 | 135.1 | 7.03 | 2.321 | 134.1 |
| 2 | 6.56 | 2.563 | 111.2 | 7.03 | 2.820 | 108.1 | |
| 3 | 5.66 | 2.178 | 149.1 | 6.70 | 2.167 | 148.8 | |
| 4 | 5.66 | 2.477 | 126.0 | 6.70 | 2.479 | 126.2 | |
| 5 | 7.29 | 1.893 | 159.9 | 7.26 | 1.968 | 159.8 | |
| RMSD (EHB) | RMSD (rHB) | RMSD (φHB) |
|---|---|---|
| 0.77 | 0.121 | 3.7 |
Appendix B.3. Analysis

Appendix C. QM/MM Liquid Water


References
- Warshel, A.; Levitt, M. Theoretical Studies of Enzymatic Reactions: Dielectric, Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Lin, H.; Truhlar, D.G. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef] [PubMed]
- Bulo, R.E.; Michel, C.; Fleurat-Lessard, P.; Sautet, P.; Heyden, A.; Lin, H.; Truhlar, D.G.; Pezeshki, S.; Lin, H.; Riahi, S.; et al. Multiscale Modeling of Chemistry in Water: Are We There Yet? J. Chem. Theory Comput. 2013, 9, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Duster, A.W.; Wang, C.H.; Garza, C.M.; Miller, D.E.; Lin, H. Adaptive quantum/molecular mechanics: What have we learned, where are we, and where do we go from here? WIRES Comput. Mol. Sci. 2017, 7, e1310. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Khorshidi, A.; Kastlunger, G.; Peterson, A.A. The potential for machine learning in hybrid QM/MM calculations. J. Chem. Phys. 2018, 148, 241740. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, C.; Reinholdt, P.; Nørby, M.S.; Kongsted, J.; Olsen, J.M.H. Response properties of embedded molecules through the polarizable embedding model. Int. J. Quantum Chem. 2018, 0, e25717. [Google Scholar] [CrossRef]
- Morzan, U.N.; Alonso de Armiño, D.J.; Foglia, N.O.; Ramírez, F.; González Lebrero, M.C.; Scherlis, D.A.; Estrin, D.A. Spectroscopy in Complex Environments from QM–MM Simulations. Chem. Rev. 2018, 118, 4071–4113. [Google Scholar] [CrossRef] [PubMed]
- Dohn, A.O.; Jónsson, E.O.; Kjær, K.S.; van Driel, T.B.; Nielsen, M.M.; Jacobsen, K.W.; Henriksen, N.E.; Møller, K.B. Direct Dynamics Studies of a Binuclear Metal Complex in Solution: The Interplay Between Vibrational Relaxation, Coherence, and Solvent Effects. J. Phys. Chem. Lett. 2014, 5, 2414–2418. [Google Scholar] [CrossRef] [PubMed]
- van Driel, T.B.; Kjær, K.S.; Hartsock, R.W.; Dohn, A.O.; Harlang, T.; Chollet, M.; Christensen, M.; Gawelda, W.; Henriksen, N.E.; Kim, J.G.; et al. Atomistic characterization of the active-site solvation dynamics of a model photocatalyst. Nat. Commun. 2016, 7, 13678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohn, A.O.; Jónsson, E.Ö.; Levi, G.; Mortensen, J.J.; Lopez-Acevedo, O.; Thygesen, K.S.; Jacobsen, K.W.; Ulstrup, J.; Henriksen, N.E.; Møller, K.B.; et al. Grid-Based Projector Augmented Wave (GPAW) Implementation of Quantum Mechanics/Molecular Mechanics (QM/MM) Electrostatic Embedding and Application to a Solvated Diplatinum Complex. J. Chem. Theory Comput. 2017, 13, 6010–6022. [Google Scholar] [CrossRef] [PubMed]
- Erba, A.; Baima, J.; Bush, I.; Orlando, R.; Dovesi, R. Large-Scale Condensed Matter DFT Simulations: Performance and Capabilities of the CRYSTAL Code. J. Chem. Theory Comput. 2017, 13, 5019–5027. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Olando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual; University of Torino: Torino, Italy, 2017. [Google Scholar]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Labat, F.; Baranek, P.; Domain, C.; Minot, C.; Adamo, C. Density functional theory analysis of the structural and electronic properties of TiO2 rutile and anatase polytypes: Performances of different exchange-correlation functionals. J. Chem. Phys. 2007, 126, 154703. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Mora-Seró, I.; De Angelis, F.; Bisquert, J.; Wang, P. Titanium Dioxide Nanomaterials for Photovoltaic Applications. Chem. Rev. 2014, 114, 10095–10130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wang, X.; Jia, Y.; Chen, X.; Han, H.; Li, C. Titanium Dioxide-based Nanomaterials for Photocatalytic Fuel Generations. Chem. Rev. 2014, 114, 9987–10043. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Rajh, T.; Dimitrijevic, N.M.; Bissonnette, M.; Koritarov, T.; Konda, V. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 10177–10216. [Google Scholar] [CrossRef] [PubMed]
- Diebold, U. Perspective: A Controversial Benchmark System for Water-oxide Interfaces: H2O/TiO2(110). J. Chem. Phys. 2017, 147, 040901. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Zhao, Z.j.; Dohnálek, Z.; Gong, J. Structural Motifs of Water on Metal Oxide Surfaces. Chem. Soc. Rev. 2017, 46, 1785–1806. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Di Valentin, C.; Fantacci, S.; Vittadini, A.; Selloni, A. Theoretical Studies on Anatase and Less Common TiO2 Phases: Bulk, Surfaces, and Nanomaterials. Chem. Rev. 2014, 114, 9708–9753. [Google Scholar] [CrossRef] [PubMed]
- Fazio, G.; Ferrighi, L.; Di Valentin, C. Spherical versus Faceted Anatase TiO2 Nanoparticles: A Model Study of Structural and Electronic Properties. J. Phys. Chem. C 2015, 119, 20735–20746. [Google Scholar] [CrossRef]
- Selli, D.; Fazio, G.; Di Valentin, C. Modelling Realistic TiO2 Nanospheres: A Benchmark Study of SCC-DFTB against DFT. J. Chem. Phys. 2017, 147, 164701. [Google Scholar] [CrossRef] [PubMed]
- Selli, D.; Fazio, G.; Di Valentin, C. Using Density Functional Theory to Model Realistic TiO2 Nanoparticles, Their Photoactivation and Interaction with Water. Catalysts 2017, 7, 357. [Google Scholar] [CrossRef]
- Shirai, K.; Fazio, G.; Sugimoto, T.; Selli, D.; Ferraro, L.; Watanabe, K.; Haruta, M.; Ohtani, B.; Kurata, H.; Di Valentin, C.; et al. Water-Assisted Hole Trapping at Highly Curved Surface of Nano-TiO2 Photocatalyst. J. Am. Chem. Soc. 2018, 140, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, L.; Boerio-Goates, J.; Woodfield, B.F. High Purity Anatase TiO2 Nanocrystals: Near Room-Temperature Synthesis, Grain Growth Kinetics, and Surface Hydration Chemistry. J. Am. Chem. Soc. 2005, 127, 8659–8666. [Google Scholar] [CrossRef] [PubMed]
- Fazio, G.; Selli, D.; Seifert, G.; Di Valentin, C. Curved TiO2 Nanoparticles in Water: Short (Chemical) and Long (Physical) Range Interfacial Effects. ACS Appl. Mater. Interfaces 2018, 10, 29943–29953. [Google Scholar] [CrossRef] [PubMed]
- Bahn, S.R.; Jacobsen, K.W. An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 2002, 4, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.; Mortensen, J.; Blomqvist, J.; Castelli, I.; Christensen, R.; Dulak, M.; Friis, J.; Groves, M.; Hammer, B.; Hargus, C.; et al. The Atomic Simulation Environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.; Sanchez, V.M.; Scherlis, D.A. A quantum-mechanics molecular-mechanics scheme for extended systems. J. Phys. Condens. Matter 2016, 28, 335201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laio, A.; VandeVondele, J.; Rothlisberger, U. A Hamiltonian Electrostatic Coupling Scheme for Hybrid Car–Parrinello Molecular Dynamics Simulations. J. Chem. Phys. 2002, 116, 6941–6947. [Google Scholar] [CrossRef]
- Freindorf, M.; Shao, Y.; Furlani, T.R.; Kong, J. Lennard–Jones parameters for the combined QM/MM method using the B3LYP/6-31G*/AMBER potential. J. Comput. Chem. 2005, 26, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Gillan, M.J.; Alfè, D.; Michaelides, A. Perspective: How good is DFT for water? J. Chem. Phys. 2016, 144, 130901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head-Gordon, T.; Hura, G. Water Structure from Scattering Experiments and Simulation. Chem. Rev. 2002, 102, 2651–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorova, T.; Seitsonen, A.P.; Hutter, J.; Kuo, I.W.; Mundy, C.J. Molecular Dynamics Simulation of Liquid Water: Hybrid Density Functionals. J. Phys. Chem. B 2006, 110, 3685–3691. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, A.P.; Bryk, T. Melting temperature of water: DFT-based molecular dynamics simulations with D3 dispersion correction. Phys. Rev. B 2016, 94, 184111. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an Improved Four-Site Water Model for Biomolecular Simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Wikfeldt, K.T.; Batista, E.R.; Vila, F.D.; Jónsson, H. A transferable H2O interaction potential based on a single center multipole expansion: SCME. Phys. Chem. Chem. Phys. 2013, 15, 16542–16556. [Google Scholar] [CrossRef] [PubMed]
- Medders, G.R.; Babin, V.; Paesani, F. Development of a “First-Principles” Water Potential with Flexible Monomers. III. Liquid Phase Properties. J. Chem. Theory Comput. 2014, 10, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, G.A.; Wikfeldt, K.T.; Ojamäe, L.; Lu, J.; Xu, Y.; Torabifard, H.; Bartók, A.P.; Csányi, G.; Molinero, V.; Paesani, F. Modeling Molecular Interactions in Water: From Pairwise to Many-Body Potential Energy Functions. Chem. Rev. 2016, 116, 7501–7528. [Google Scholar] [CrossRef] [PubMed]
- Babin, V.; Leforestier, C.; Paesani, F. Development of a “First Principles” Water Potential with Flexible Monomers: Dimer Potential Energy Surface, VRT Spectrum, and Second Virial Coefficient. J. Chem. Theory Comput. 2013, 9, 5395–5403. [Google Scholar] [CrossRef] [PubMed]
- Jurečka, P.; Šponer, J.; Černý, J.; Hobza, P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Rajh, T.; Chen, L.X.; Lukas, K.; Liu, T.; Thurnauer, M.C.; Tiede, D.M. Surface Restructuring of Nanoparticles: An Efficient Route for Ligand-Metal Oxide Crosstalk. J. Phys. Chem. B 2002, 106, 10543–10552. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.; Roetti, C. Crystal-field effects on the topological properties of the electron-density in molecular-crystals. The case of urea. J. Chem. Phys. B 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bates, D.M.; Tschumper, G.S. CCSD(T) Complete Basis Set Limit Relative Energies for Low-Lying Water Hexamer Structures. J. Phys. Chem. A 2009, 113, 3555–3559. [Google Scholar] [CrossRef] [PubMed]
- Temelso, B.; Archer, K.A.; Shields, G.C. Benchmark Structures and Binding Energies of Small Water Clusters with Anharmonicity Corrections. J. Phys. Chem. A 2011, 115, 12034–12046. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Walser, R.; Mark, A.E.; van Gunsteren, W.F. On the Temperature and Pressure Dependence of a Range of Properties of a Type of Water Model Commonly Used in High-Temperature Protein Unfolding Simulations. Biophys. J. 2000, 78, 2752–2760. [Google Scholar] [CrossRef] [Green Version]
- Schwegler, E.; Grossman, J.C.; Gygi, F.; Galli, G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. II. J. Chem. Phys. 2004, 121, 5400–5409. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.C.; Schwegler, E.; Draeger, E.W.; Gygi, F.; Galli, G. Towards an assessment of the accuracy of density functional theory for first principles simulations of water. J. Chem. Phys. 2004, 120, 300–311. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







| Type | σOO (Å) | ϵOO (kcal/mol) |
|---|---|---|
| TIP3P | 3.15061 | 0.1521 |
| Dimer Fit (DF) | 3.89048 | 0.0122 |
| Cluster Fit (CF) | 3.10031 | 0.2629 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ougaard Dohn, A.; Selli, D.; Fazio, G.; Ferraro, L.; Mortensen, J.J.; Civalleri, B.; Di Valentin, C. Interfacing CRYSTAL/AMBER to Optimize QM/MM Lennard–Jones Parameters for Water and to Study Solvation of TiO2 Nanoparticles. Molecules 2018, 23, 2958. https://doi.org/10.3390/molecules23112958
Ougaard Dohn A, Selli D, Fazio G, Ferraro L, Mortensen JJ, Civalleri B, Di Valentin C. Interfacing CRYSTAL/AMBER to Optimize QM/MM Lennard–Jones Parameters for Water and to Study Solvation of TiO2 Nanoparticles. Molecules. 2018; 23(11):2958. https://doi.org/10.3390/molecules23112958
Chicago/Turabian StyleOugaard Dohn, Asmus, Daniele Selli, Gianluca Fazio, Lorenzo Ferraro, Jens Jørgen Mortensen, Bartolomeo Civalleri, and Cristiana Di Valentin. 2018. "Interfacing CRYSTAL/AMBER to Optimize QM/MM Lennard–Jones Parameters for Water and to Study Solvation of TiO2 Nanoparticles" Molecules 23, no. 11: 2958. https://doi.org/10.3390/molecules23112958
APA StyleOugaard Dohn, A., Selli, D., Fazio, G., Ferraro, L., Mortensen, J. J., Civalleri, B., & Di Valentin, C. (2018). Interfacing CRYSTAL/AMBER to Optimize QM/MM Lennard–Jones Parameters for Water and to Study Solvation of TiO2 Nanoparticles. Molecules, 23(11), 2958. https://doi.org/10.3390/molecules23112958