Conversion of Medium-Sized Lactams to α-Vinyl or α-Acetylenyl Azacycles via N,O-Acetal TMS Ethers

 , and
, and
Abstract
: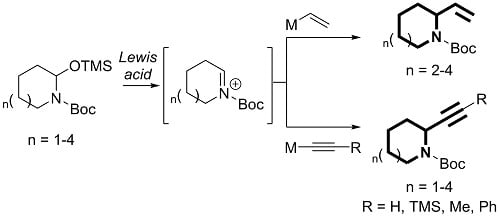
1. Introduction
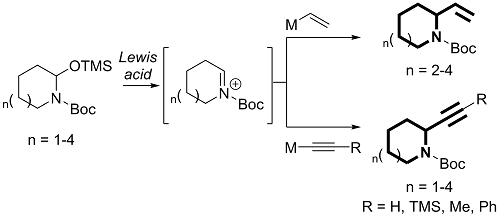
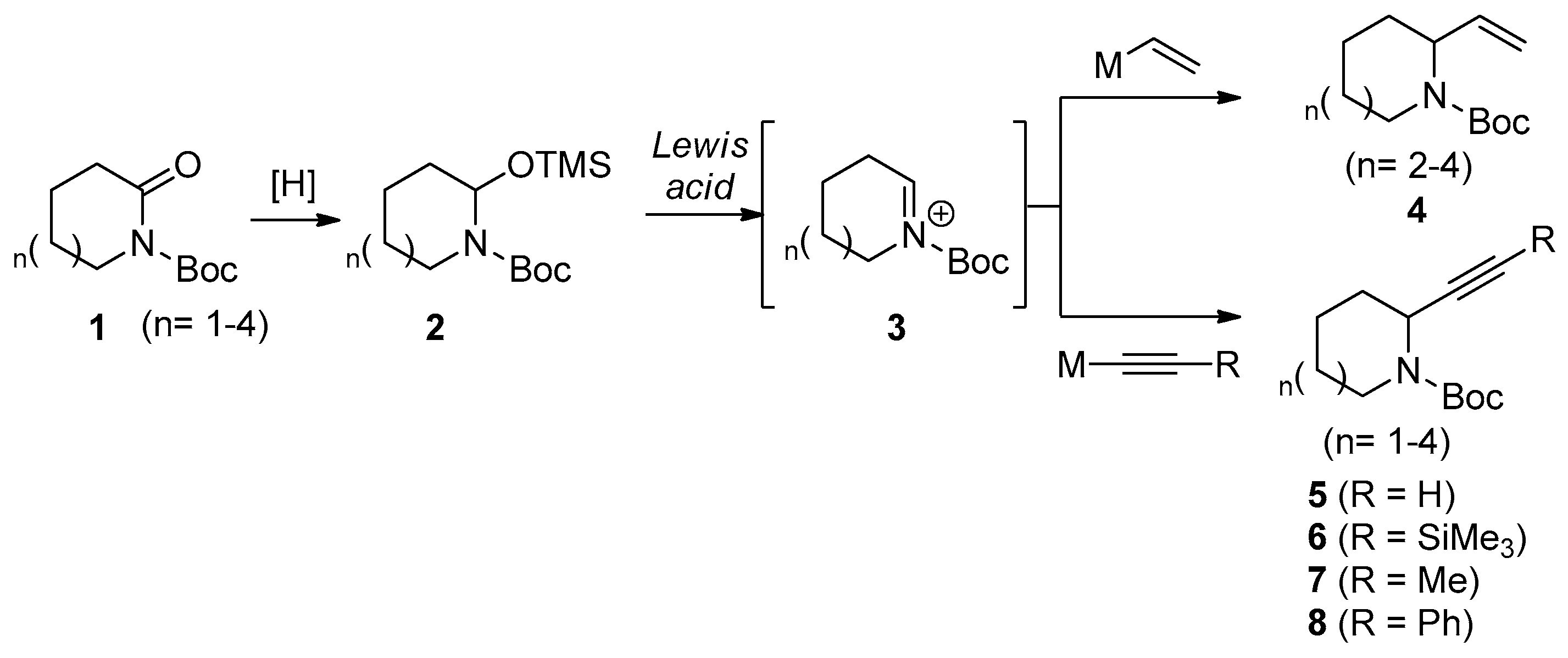
2. Results and Discussion
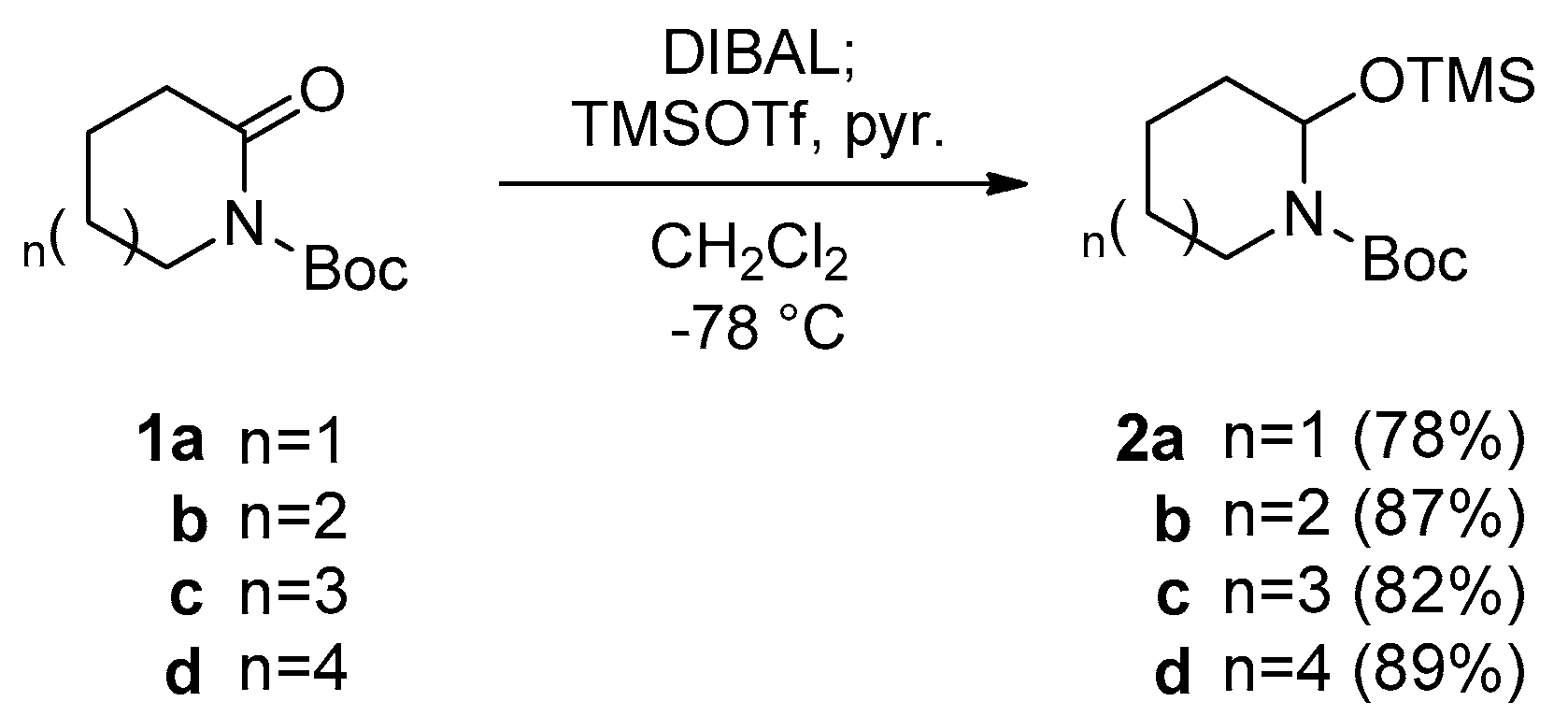
2.1. Preparation of N,O-Acetal TMS Ether
2.2. Optimization of α-Vinylation
2.3. Optimization α-Acetylenylation
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Representative Procedure: Synthesis of tert-Butyl 2-vinylazonane-1-carboxylate (4d)
3.2.2. Representative Procedure: Synthesis of tert-Butyl 2-ethynylazonane-1-carboxylate (5d)
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Wu, P.; Nielsen, T.E. Scaffold diversity from N-acyliminium ions. Chem. Rev. 2017, 117, 7811–7856. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Cai, C.; Mayorov, A.V.; Xiong, C.; Cabello, M.; Soloshonok, V.A.; Swift, J.R.; Trivedi, D.; Hruby, V.J. Biological and conformational study of beta-substituted prolines in MT-II template: Steric effects leading to human MC5 receptor selectivity. J. Pept. Res. 2004, 63, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Takahata, H.; Takahashi, K.; Wang, E.-C.; Yamazaki, T. Alkynylation of thiolactams: New synthesis of α-substituted pyrrolidine and piperidine alkaloids. J. Chem. Soc. Perkin Trans. 1989, 1, 1211–1214. [Google Scholar] [CrossRef]
- Cook, G.R.; Beholz, L.G.; Stille, J.R. Construction of hydroxylated alkaloids (±)-mannonolactam,(±)-deoxymannojirimycin, and (±)-prosopinine through aza-annulation. J. Org. Chem. 1994, 59, 3575–3584. [Google Scholar] [CrossRef]
- Schär, P.; Renaud, P. Total synthesis of the marine alkaloid (±)-lepadiformine via a radical carboazidation. Org. Lett. 2006, 8, 1569–1571. [Google Scholar] [CrossRef] [PubMed]
- Neipp, C.E.; Martin, S.F. Synthesis of bridged azabicyclic structures via ring-closing olefin metathesis. J. Org. Chem. 2003, 68, 8867–8878. [Google Scholar] [CrossRef] [PubMed]
- Jaekel, M.; Qu, J.; Schnitzer, T.; Helmchen, G. Addition of Organometallic Reagents to Chiral N-methoxylactams: Enantioselective syntheses of pyrrolidines and piperidines. Chem. Eur. J. 2013, 19, 16746–16755. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, L.; Meyers, A. Electrophilic formamidines. Organometallic addition to 2-methoxy pyrrolidine or piperidine N-t-butyl formamidines. Formation of 2-substituted pyrrolidines and piperidines. Tetrahedron Lett. 1990, 31, 4723–4726. [Google Scholar] [CrossRef]
- Ha, J.D.; Cha, J.K. Total synthesis of clavepictines a and b diastereoselective cyclization of δ-aminoallenes. J. Am. Chem. Soc. 1999, 121, 10012–10020. [Google Scholar] [CrossRef]
- Jiang, J.; DeVita, R.J.; Doss, G.A.; Goulet, M.T.; Wyvratt, M.J. Asymmetric synthesis of chiral, nonracemic trifluoromethyl-substituted piperidines and decahydroquinolines. J. Am. Chem. Soc. 1999, 121, 593–594. [Google Scholar] [CrossRef]
- Batey, R.A.; MacKay, D.B.; Santhakumar, V. Alkenyl and aryl boronates mild nucleophiles for the stereoselective formation of functionalized n-heterocycles. J. Am. Chem. Soc. 1999, 121, 5075–5076. [Google Scholar] [CrossRef]
- Itoh, T.; Yamazaki, N.; Kibayashi, C. asymmetric synthesis of (−)-adaline. Org. Lett. 2002, 4, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
- Collado, I.; Ezquerra, J.; Pedregal, C. Stereoselective addition of Grignard-derived organocopper reagents to N-acyliminium ions: Synthesis of enantiopure 5- and 4,5-substituted prolinates. J. Org. Chem. 1995, 60, 5011–5015. [Google Scholar] [CrossRef]
- Jung, J.W.; Kim, S.H.; Suh, Y.G. Advances in aza-claisen-rearrangement-induced ring-expansion strategies. Asian J. Org. Chem. 2017, 6, 1117–1129. [Google Scholar] [CrossRef]
- Campos, K.R. Direct sp3 C—H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev. 2007, 36, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.; MacMillan, D.W. Photoredox α-vinylation of α-amino acids and N-aryl amines. J. Am. Chem. Soc. 2014, 136, 11602–11605. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Kim, S.-H.; Jung, J.-K.; Shin, D.-Y. The versatile conversion of lactams to the α-alkylated azacycles via cyclic N,O-acetal TMS ether. Tetrahedron Lett. 2002, 43, 3165–3167. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Lee, Y.-S.; Kim, S.-H.; Jung, J.-K.; Yun, H.; Jang, J.; Kim, N.-J.; Jung, J.-W. A stereo-controlled access to functionalized macrolactams via an aza-Claisen rearrangement. Org. Biomol. Chem. 2012, 10, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.W.; Shin, D.Y.; Seo, S.Y.; Kim, S.H.; Paek, S.M.; Jung, J.K.; Suh, Y.G. A new entry to functionalized cycloalkylamines: Diastereoselective intramolecular amidoalkylation of N,O-acetal TMS ether possessing allylsilane. Tetrahedron Lett. 2005, 46, 573–575. [Google Scholar] [CrossRef]
- Meana, I.; Albeniz, A.C.; Espinet, P. Selective green coupling of alkynyltins and allylic halides to trienynes via a tandem double stille reaction. Adv. Synth. Catal. 2010, 352, 2887–2891. [Google Scholar] [CrossRef]
- Wu, X.; Cruz, F.A.; Lu, A.; Dong, V.M. Tandem catalysis: Transforming alcohols to alkenes by oxidative dehydroxymethylation. J. Am. Chem. Soc. 2018, 140, 10126–10130. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}

| Entry a | n | M | Additive | Lewis Acid | Solvent | Yield b |
|---|---|---|---|---|---|---|
| 1 | 4 | Li | BF3·OEt2 | THF | ND c | |
| 2 | 4 | SiMe3 | BF3·OEt2 | CH2Cl2 | ND c | |
| 3 | 4 | MgBr | BF3·OEt2 | THF | ND c | |
| 4 | 4 | MgBr | CuBr·SMe2 | BF3·OEt2 | THF | 63 |
| 5 | 4 | MgBr | CuBr·SMe2 | BF3·OEt2 | Et2O | 32 |
| 6 | 4 | MgBr | CuI | BF3·OEt2 | THF | ND c |
| 7 | 4 | MgBr | CuOTf | BF3·OEt2 | THF | ND c |
| 8 | 3 | MgBr | CuBr·SMe2 | BF3·OEt2 | THF | 60 |
| 9 | 2 | MgBr | CuBr·SMe2 | BF3·OEt2 | THF | 65 |
| 10 | 1 | MgBr | CuBr·SMe2 | BF3·OEt2 | THF | ND c |

| Entry a | M | Additive | Lewis acid | Solvent | Yield b |
|---|---|---|---|---|---|
| 1 | MgBr | CuBr·SMe2 | BF3·OEt2 | THF | ND c |
| 2 | MgBr | BF3·OEt2 | THF | ND c | |
| 3 | Li | BF3·OEt2 | THF | ND c | |
| 4 | SiMe3 | BF3·OEt2 | CH2Cl2 | ND c | |
| 5 | n-Bu3Sn | BF3·OEt2 | CH2Cl2 | 58 | |
| 6 | n-Bu3Sn | BF3·OEt2 | THF | ND c | |
| 7 | n-Bu3Sn | BF3·OEt2 | PhCH3 | 37 | |
| 8 | n-Bu3Sn | BF3·OEt2 | Et2O | 29 | |
| 9 | n-Bu3Sn | SnCl4 | CH2Cl2 | ND c | |
| 10 | n-Bu3Sn | TiCl4 | CH2Cl2 | ND c | |
| 11 | n-Bu3Sn | TMSOTf | CH2Cl2 | 46 |

| Entry a | n | R | Yield b |
|---|---|---|---|
| 1 | 1 | H | 30 |
| 2 | 1 | SiMe3 | 29 |
| 3 | 1 | Me | 35 |
| 4 | 1 | Ph | 31 |
| 5 | 2 | H | 28 |
| 6 | 2 | SiMe3 | 38 |
| 7 | 2 | Me | 89 |
| 8 | 2 | Ph | 88 |
| 9 | 3 | H | 84 |
| 10 | 3 | SiMe3 | 36 |
| 11 | 3 | Me | 82 |
| 12 | 3 | Ph | 68 |
| 13 | 4 | H | 58 |
| 14 | 4 | SiMe3 | 64 |
| 15 | 4 | Me | 80 |
| 16 | 4 | Ph | 81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Jang, J.; Choi, G.; Chung, S.; Lim, C.; Hur, J.; Kim, H.S.; Na, Y.; Son, W.S.; Suh, Y.-G.; et al. Conversion of Medium-Sized Lactams to α-Vinyl or α-Acetylenyl Azacycles via N,O-Acetal TMS Ethers. Molecules 2018, 23, 3023. https://doi.org/10.3390/molecules23113023
Kim M, Jang J, Choi G, Chung S, Lim C, Hur J, Kim HS, Na Y, Son WS, Suh Y-G, et al. Conversion of Medium-Sized Lactams to α-Vinyl or α-Acetylenyl Azacycles via N,O-Acetal TMS Ethers. Molecules. 2018; 23(11):3023. https://doi.org/10.3390/molecules23113023
Chicago/Turabian StyleKim, Minjun, Jaebong Jang, Goyoung Choi, Sungkyun Chung, Changjin Lim, Joonseong Hur, Hyun Su Kim, Younghwa Na, Woo Sung Son, Young-Ger Suh, and et al. 2018. "Conversion of Medium-Sized Lactams to α-Vinyl or α-Acetylenyl Azacycles via N,O-Acetal TMS Ethers" Molecules 23, no. 11: 3023. https://doi.org/10.3390/molecules23113023
APA StyleKim, M., Jang, J., Choi, G., Chung, S., Lim, C., Hur, J., Kim, H. S., Na, Y., Son, W. S., Suh, Y.-G., Jung, J.-W., & Kim, S.-H. (2018). Conversion of Medium-Sized Lactams to α-Vinyl or α-Acetylenyl Azacycles via N,O-Acetal TMS Ethers. Molecules, 23(11), 3023. https://doi.org/10.3390/molecules23113023