Gelation of Poly(Vinylidene Fluoride) Solutions in Native and Organically Modified Silica Nanopores



Abstract
:1. Introduction
2. Results and Discussion
2.1. FTIR and XRD Characterization
2.2. DSC Data on Gelation of the Bulk Systems
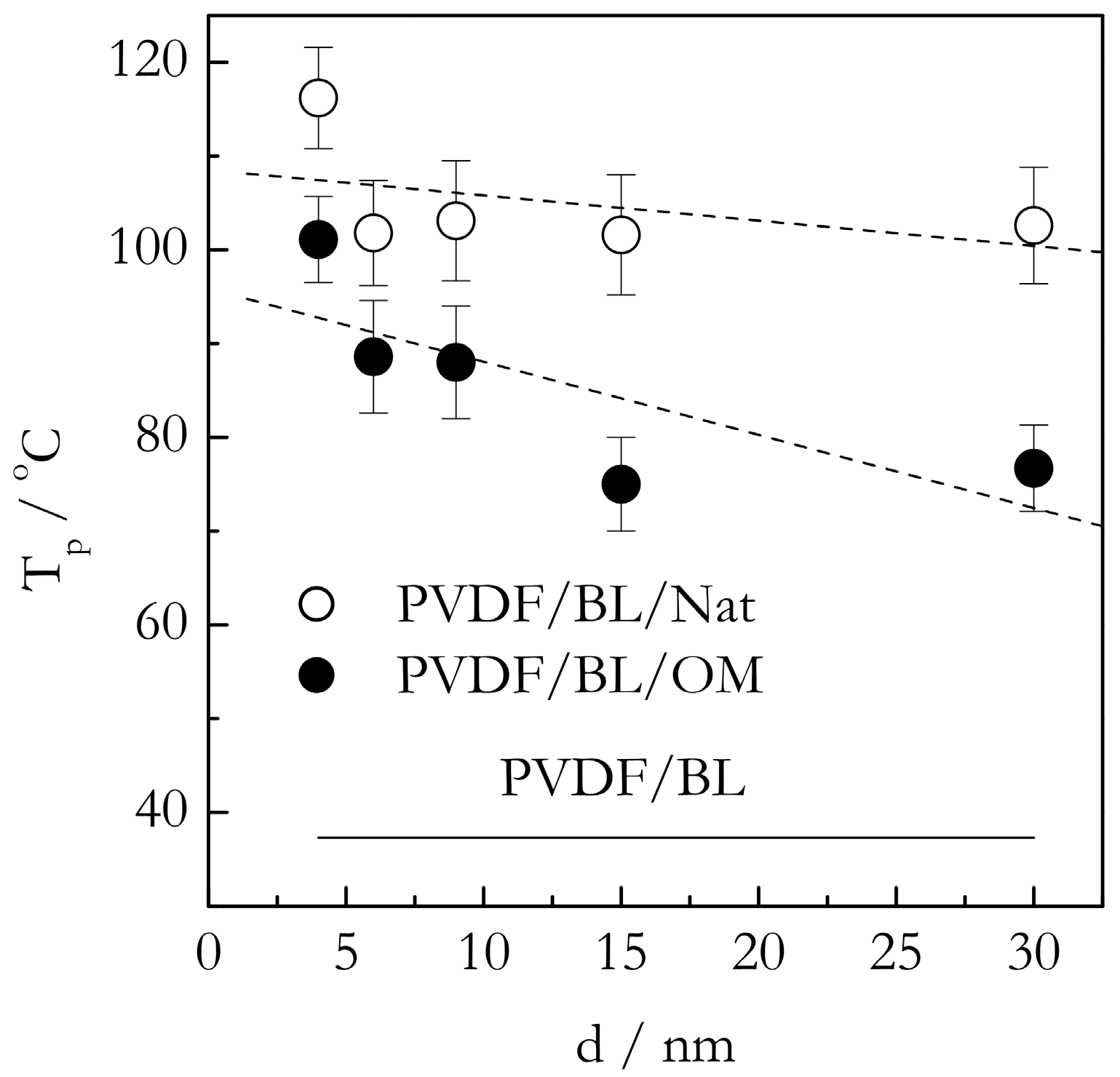
2.3. DSC Data on Gelation of Nanoconfined Systems
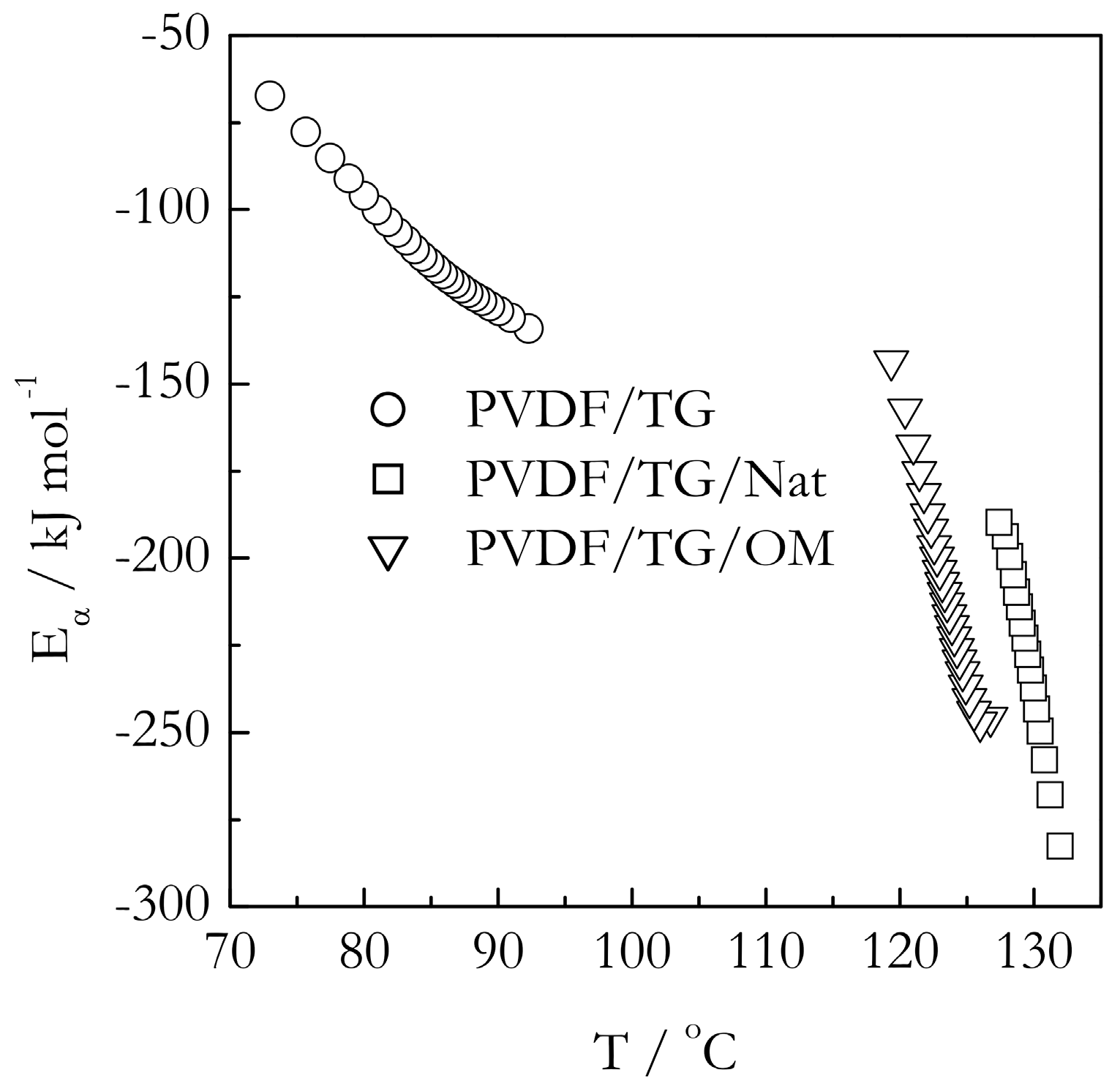
2.4. Kinetics of Gelation
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vinogradov, S.V. Hydrophilic colloidal networks (micro- and nanogels) in drug delivery and discovery. In Structure and Functional Properties of Colloidal Systems, 1st ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Molina, M.; Asadian-Birjand, M.; Balach, J.; Bergueiro, J.; Miceli, E.; Calderon, M. Stimuli-responsive nanogel composites and their application in nanomedicine. Chem. Soc. Rev. 2015, 44, 6161–6186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, K.S.; Desale, S.S.; Bronich, T.K. Nanogels: An overview of properties, biomedical applications and obstacles to clinical translation. J. Control. Release 2016, 240, 109–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.K.; Drumrighta, R.; Siegwartb, D.J.; Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar] [CrossRef]
- Vinogradov, S.V.; Bronich, T.K.; Kabanov, A.V. Nanosized cationic hydrogels for drug delivery: Preparation, properties and interactions with cells. Adv. Drug Deliv. Rev. 2002, 54, 135–147. [Google Scholar] [CrossRef]
- Prado, J.R.; Chen, J.; Kharlampieva, E.; Vyazovkin, S. Melting of gelatin gels confined to silica nanopores. Phys. Chem. Chem. Phys. 2016, 18, 29056–29063. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Q.; Zhou, S. Carbon-based hybrid nanogels: A synergistic nanoplatform for combined biosensing, bioimaging, and responsive drug delivery. Chem. Soc. Rev. 2018, 47, 4198–4232. [Google Scholar] [CrossRef] [PubMed]
- Giesa, T.; Buehler, M.J. Nanoconfinement and the strength of biopolymers. Annu. Rev. Biophys. 2013, 42, 651–673. [Google Scholar] [CrossRef] [PubMed]
- Carroll, N.J.; Rathod, S.B.; Derbins, E.; Mendez, S.; Weitz, D.A.; Petsev, D.N. Droplet-Based Microfluidics for Emulsion and Solvent Evaporation Synthesis of Monodisperse Mesoporous Silica Microspheres. Langmuir 2008, 24, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S. Mesoporous Silica Nanoparticles for Drug Delivery and Biosensing Applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Motornov, M.; Roiter, Y.; Tokarev, I.; Minko, S. Stimuli-responsive nanoparticles, nanogels and capsules for integrated multifunctional intelligent systems. Prog. Polym. Sci. 2010, 35, 174–211. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry of Silica; Wiley Inter-Science Publication, John Wiley & Sons, Inc.: New York, NY, USA, 1979. [Google Scholar]
- Hossain, D.; Pittman, C.U.; Saebo, S. Structures, Stabilities, and Electronic Properties of Endo- and Exohedral Complexes of T10−Polyhedral Oligomeric Silsesquioxane Cages. J. Phys. Chem. C 2007, 111, 6199–6209. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Prado, J.R.; Vyazovkin, S. Phase separation of triethylamine and water in native and organically modified silica nanopores. J. Chem. Phys. 2017, 147, 114508. [Google Scholar] [CrossRef] [PubMed]
- Domszy, R.C.; Alamo, R.; Edwards, C.O.; Mandelkern, L. Thermoreversible Gelation and Crystallization of Homopolymers and Copolymers. Macromolecules 1986, 19, 310–325. [Google Scholar] [CrossRef]
- Guenet, J.-M. Thermoreversible Gelation of Polymers and Biopolymers; Academic Press: London, UK, 1992. [Google Scholar]
- Farasat, R.; Vyazovkin, S. Nanoconfined Solid–Solid Transitions: Attempt To Separate the Size and Surface Effects. J. Phys. Chem. C 2015, 119, 9627–9636. [Google Scholar] [CrossRef]
- Haro-Perez, C.; Garcia-Castillo, A.; Arauz-Lara, J.L. Confinement-induced fluid-gel transition in polymeric solutions. Langmuir 2009, 25, 8911–8914. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, Y.; Lee, C.H.; Shen, A.Q. Confinement Effects on the Self-Assembly of 1,3:2,4-Di-p-methylbenzylidene Sorbitol Based Organogel. Langmuir 2008, 24, 10432–10436. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Li, J.L.; Liu, X.Y.; Ma, Y.Q.; Wang, Y.J. Size invariance of fibrous networks of supramolecular soft materials during formation under critical volume confinement. Soft Matter 2012, 8, 5187–5193. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, W.J.; Li, J.L.; Wang, R.Y. Distinct kinetics of molecular gelation in a confined space and its relation to the structure and property of thin gel films. Phys. Chem. Chem. Phys. 2015, 17, 8258–8265. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, Z.; Feng, G.; Wang, H.; Xu, H.; Yang, X.; Yang, Y. Self-assembly of gelators confined within the nano-scale interlayer space of organo-montmorillonite. Phys. Chem. Chem. Phys. 2008, 10, 6479–6482. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baker, A.N.; Vyazovkin, S. Formation and Thermal Behavior of Polystyrene and Polystyrene-Clay Gels. Macromol. Chem. Phys. 2008, 209, 2367–2373. [Google Scholar] [CrossRef]
- Chen, K.; Wilkie, C.A.; Vyazovkin, S. Nanoconfinement Revealed in Degradation and Relaxation Studies of Two Structurally Different Polystyrene-Clay Systems. J. Phys. Chem. B 2007, 111, 12685–12692. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Vyazovkin, S. Temperature dependence of sol-gel conversion kinetics in gelatin-water system. Macromol. Biosci. 2009, 9, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baker, A.N.; Vyazovkin, S. Concentration Effect on Temperature Dependence of Gelation Rate in Aqueous Solutions of Methylcellulose. Macromol. Chem. Phys. 2009, 210, 211–216. [Google Scholar] [CrossRef]
- Dranca, I.; Vyazovkin, S. Thermal stability of gelatin gels: Effect of preparation conditions on the activation energy barrier to melting. Polymer 2009, 50, 4859–4867. [Google Scholar] [CrossRef]
- Guigo, N.; Sbirrazzuoli, N.; Vyazovkin, S. Gelation on heating of supercooled gelatin solutions. Macromol. Rapid Commun. 2012, 33, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Guigo, N.; Sbirrazzuoli, N.; Vyazovkin, S. Atypical gelation in gelatin solutions probed by ultra-fast calorimetry. Soft Matter 2012, 8, 7116–7121. [Google Scholar] [CrossRef]
- Prado, J.R.; Vyazovkin, S. Melting of Gelatin Gels Containing Laponite, Montmorillonite, and Chitosan Particles. Macromol. Chem. Phys. 2014, 215, 867–872. [Google Scholar] [CrossRef]
- Cho, J.W.; Song, H.Y.; Kim, S.Y. Thermoreversible gelation of poly(vinylidene fluoride) in γ-butyrolactone solution. Polymer 1993, 34, 1024–1027. [Google Scholar] [CrossRef]
- Mal, S.; Nandi, A.K. A Thermodynamic Study on the Thermoreversible Poly(vinylidene fluoride) Gels in Acetophenone, Ethyl Benzoate, and Glyceryl Tributyrate. Langmuir 1998, 14, 2238–2244. [Google Scholar] [CrossRef]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer: Berlin, Germany, 2005; p. 894. [Google Scholar]
- Cai, X.; Tingping, L.; Sun, D.; Lin, L. A critical analysis of the α, β and γ phases in poly(vinylidene fluoride) using FTIR. RSC Adv. 2017, 7, 15382–15389. [Google Scholar] [CrossRef] [Green Version]
- Okabe, M.; Wada, R.; Tazaki, M.; Homma, T. The Flory−Huggins Interaction Parameter and Thermoreversible Gelation of Poly(vinylidene fluoride) in Organic Solvents. Polym. J. 2003, 35, 798–803. [Google Scholar] [CrossRef]
- Jurczuk, K.; Galeski, A.; Mackey, M.; Hiltner, A.; Baer, E. Orientation of PVDF alpha and gamma crystals in nanolayered films. Colloid Polym. Sci. 2015, 293, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Wallmark, I.; Krackov, M.H.; Chu, S.-H.; Mautner, H.G. Effects of replacing either of the oxygens of the ester group by sulfur and selenium. Investigations of the sulfur and selenium isologs of gamma.-butyrolactone and of phthalide. J. Am. Chem. Soc. 1970, 92, 4447–4450. [Google Scholar] [CrossRef]
- Voice, A.M.; Southall, J.P.; Rogers, V.; Matthews, K.H.; Davies, G.R.; McIntyreI, J.E.; Ward, M. Thermoreversible polymer gel electrolytes. Polymer 1994, 35, 3363–3372. [Google Scholar] [CrossRef]
- Fornefeld-Schwarz, U.M.; Svejda, P. Refractive Indices and Relative Permittivities of Liquid Mixtures of γ-Butyrolactone, γ-Valerolactone, δ-Valerolactone, or ε-Caprolactone + Benzene, + Toluene, or + Ethylbenzene at 293.15 K and 313.15 K and Atmospheric Pressure. J. Chem. Eng. Data 1999, 44, 597–604. [Google Scholar] [CrossRef]
- Moumouzias, G.; Ritzoulis, G. Relative Permittivities and Refractive Indices of γ-Butyrolactone with o-Xylene and m-Xylene. J. Chem. Eng. Data 1999, 44, 1273–1278. [Google Scholar] [CrossRef]
- Ko, K.; Ryoichi, F. The Dipole Moments of the Oligether of Ethylene Glycol. Bull. Chem. Soc. Jpn. 1966, 39, 608–610. [Google Scholar] [Green Version]
- Riadigos, C.F.; Iglesias, R.; Rivas, M.A.; Iglesias, T.P. Permittivity and density of the systems (monoglyme, diglyme, triglyme, or tetraglyme + n-heptane) at several temperatures. J. Chem. Thermodyn. 2011, 43, 275–283. [Google Scholar] [CrossRef]
- Freund, J.E. Modern Elementary Statistics, 10th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2001. [Google Scholar]
- Godard, P.; Biebuyck, J.J.; Daumerie, M.; Naveau, H.; Mercier, J.P. Crystallization and melting of aqueous gelatin. J. Polym. Sci. Polym. Phys. Ed. 1978, 16, 1817–1828. [Google Scholar] [CrossRef]
- Schultz, J.M. Polymer Crystallization: The Development of Crystalline Order in Thermoplastic Polymers; ACS: Washington, DC, USA; Oxford University Press: Washington, DC, USA, 2001. [Google Scholar]
- Mandelkern, L. Crystallization of Polymers: Kinetics and Mechanisms; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Mullin, J.W. Crystallization, 4th ed.; Butterworth-Heinemann: Oxford, UK, 2002. [Google Scholar]
- Turnbull, D.; Fisher, J.C. Rate of Nucleation in Condensed Systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Ohkura, M.; Kanaya, T.; Kaji, K. Gelation rates of poly(vinyl alcohol) solution. Polymer 1992, 33, 5044–5048. [Google Scholar] [CrossRef]
- Farasat, R.; Yancey, B.; Vyazovkin, S. High Temperature Solid−Solid Transition in Ammonium Chloride Confined to Nanopores. J. Phys. Chem. C 2013, 117, 13713–13721. [Google Scholar] [CrossRef]
- Farasat, R.; Vyazovkin, S. Coil-to-Globule Transition of Poly(N-isopropylacrylamide) in Aqueous Solution: Kinetics in Bulk and Nanopores. Macromol. Chem. Phys. 2014, 215, 2112–2118. [Google Scholar] [CrossRef]
- Vyazovkin, S. Isoconversional Kinetics of Thermally Stimulated Processes; Springer: Heildelberg, Germany, 2015. [Google Scholar]
- Vyazovkin, S. Modification of the Integral Isoconversional Method to Account for Variation in the Activation Energy. J. Comput. Chem. 2001, 22, 178–183. [Google Scholar] [CrossRef]
- Vyazovkin, S. Isoconversional Kinetics of Polymers: The Decade Past. Macromol. Rapid Commun. 2017, 38, 1600615. [Google Scholar] [CrossRef] [PubMed]
- Stanford, V.L.; McCulley, C.M.; Vyazovkin, S. Isoconversional Kinetics of Nonisothermal Crystallization of Salts from Solutions. J. Phys. Chem. B 2016, 120, 5703–5709. [Google Scholar] [CrossRef] [PubMed]
- Pace, R.J.; Datyner, A. Statistical mechanical model of diffusion of complex penetrants in polymers. II. Applications. J. Polym. Sci.: Polym. Phys. Ed. 1979, 17, 1693–1708. [Google Scholar] [CrossRef]
- Pace, R.J.; Datyner, A. Statistical mechanical model for diffusion of simple penetrants in polymers. III. Applications—Vinyl and related polymers. J. Polym. Sci.: Polym. Phys. Ed. 1979, 17, 465–476. [Google Scholar] [CrossRef]
- Bourret, A. Low-Density Silica Aerogels Observed by High-Resolution Electron Microscopy. Europhys. Lett. 1988, 6, 731–737. [Google Scholar] [CrossRef]
- Anwander, R.; Nagl, I.; Widenmeyer, M.; Engelhardt, G.; Groeger, O.; Palm, C.; Röser, T. Surface Characterization and Functionalization of MCM-41 Silicas via Silazane Silylation. J. Phys. Chem. B 2000, 104, 3532–3544. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, Greece, 1979. [Google Scholar]
- Lutringer, G.; Weill, G. Solution properties of poly(vinylidene fluoride): 1. Macromolecular characterization of soluble samples. Polymer 1991, 32, 877–883. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measured Values | |||
|---|---|---|---|
| Nominal Pore Diameter (nm) | Pore Diameter (nm) | Surface Area (m2 g−1) | Pore Volume (cm3 g−1) |
| 4 | 3.9 | 598 | 0.60 |
| 6 | 5.7 | 496 | 0.71 |
| 9 | 10.0 | 358 | 0.80 |
| 15 | 16.7 | 285 | 1.19 |
| 30 | 28.4 | 175 | 1.24 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Dzib, A.; Vyazovkin, S. Gelation of Poly(Vinylidene Fluoride) Solutions in Native and Organically Modified Silica Nanopores. Molecules 2018, 23, 3025. https://doi.org/10.3390/molecules23113025
Espinosa-Dzib A, Vyazovkin S. Gelation of Poly(Vinylidene Fluoride) Solutions in Native and Organically Modified Silica Nanopores. Molecules. 2018; 23(11):3025. https://doi.org/10.3390/molecules23113025
Chicago/Turabian StyleEspinosa-Dzib, Alejandra, and Sergey Vyazovkin. 2018. "Gelation of Poly(Vinylidene Fluoride) Solutions in Native and Organically Modified Silica Nanopores" Molecules 23, no. 11: 3025. https://doi.org/10.3390/molecules23113025
APA StyleEspinosa-Dzib, A., & Vyazovkin, S. (2018). Gelation of Poly(Vinylidene Fluoride) Solutions in Native and Organically Modified Silica Nanopores. Molecules, 23(11), 3025. https://doi.org/10.3390/molecules23113025