
Function and Interactions of ERCC1-XPF in DNA Damage Response


Abstract
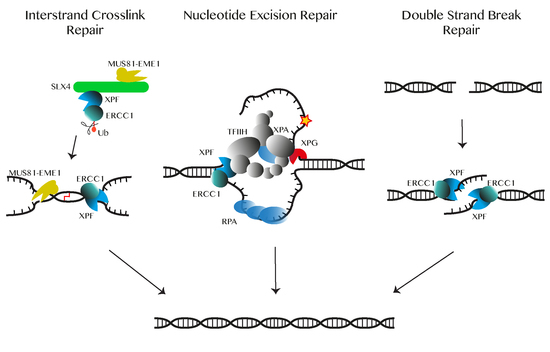
:
{kind=link}
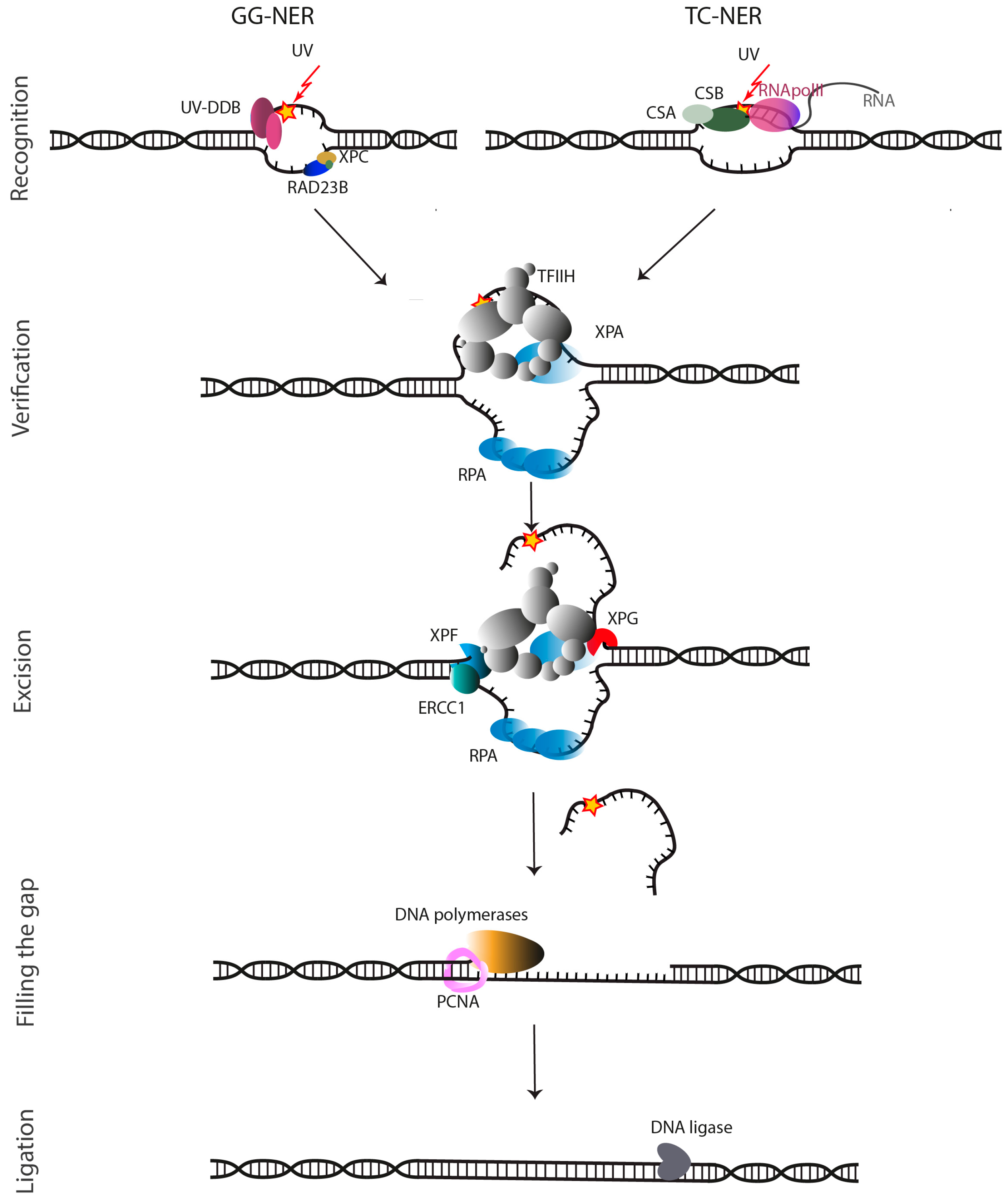{kind=link}
{kind=link}
{kind=link}
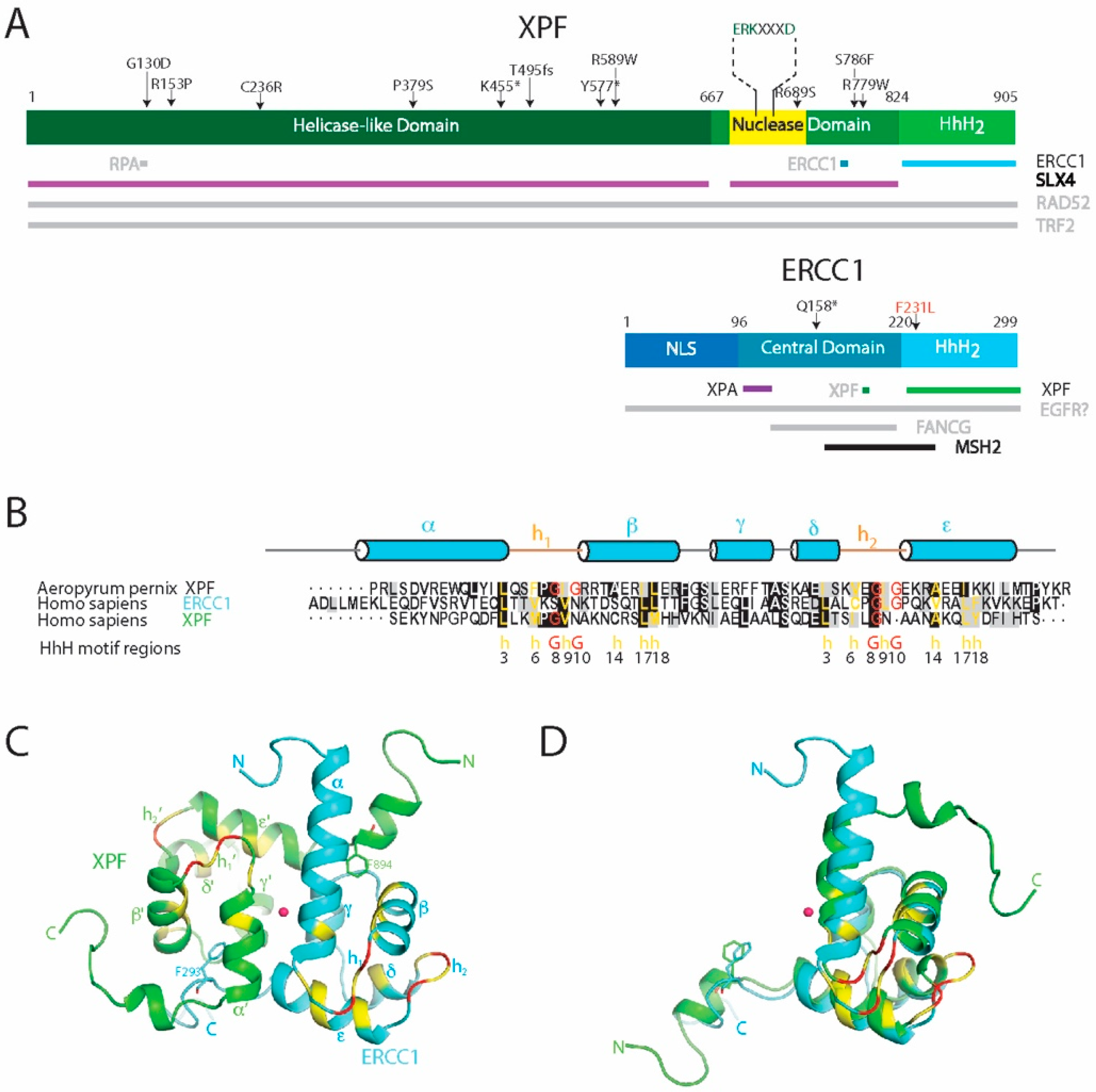{kind=link}
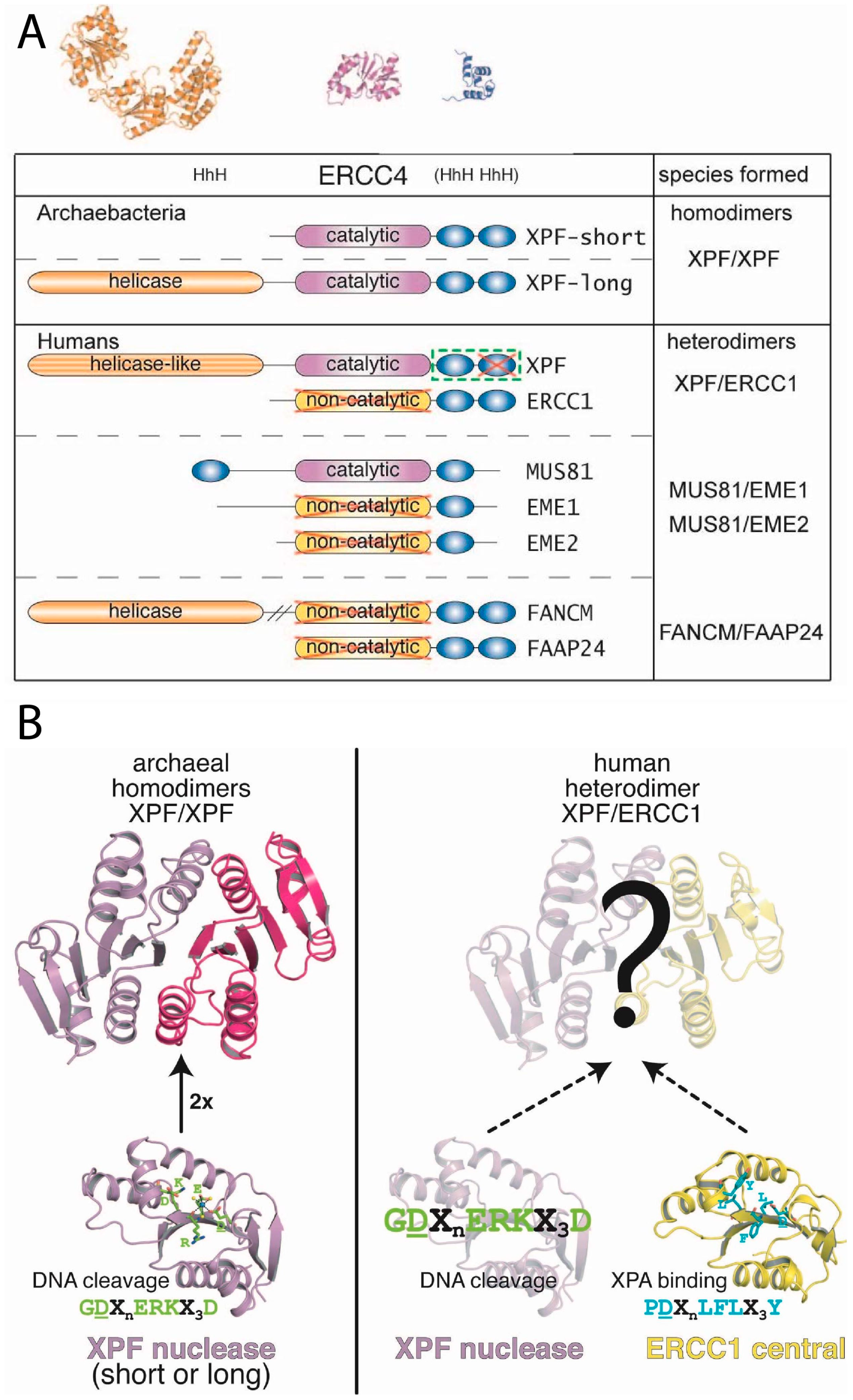{kind=link}
1. Introduction
2. Function of ERCC1-XPF in DNA Repair Pathways
2.1. Nucleotide Excision Repair (NER)
2.2. Double-Strand Break Repair
2.2.1. Homologous Recombination (HR)
2.2.2. Non-Homologous End Joining (NHEJ)
2.2.3. Microhomology Mediated End-Joining (MMEJ)
2.3. Single-Strand Break Repair
2.4. Interstrand Cross-Link (ICL) Repair
2.4.1. ICL Replication/Recombination-Dependent Pathway
2.4.2. ICL Replication-Independent Repair Pathway
2.4.3. Other ICL Repair Pathways
2.5. Roles of ERCC1-XPF Beyond DNA Repair
3. Physiological Consequences of Impaired ERCC1-XPF
3.1. Xeroderma Pigmentosum (XP)
3.2. Cockayne Syndrome
3.3. Fanconi Anemia Disorder
4. ERCC1 and XPF Belong to the XPF Nuclease Family
4.1. The Superfamily 2 Helicase-Like Domain
4.2. The ERCC4 Endonuclease Domain
4.3. The ERCC1 Central Domain of ERCC1
4.4. The Tandem Helix-Hairpin-Helix Domain (HhH)2
5. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2015; pp. 1–1342. [Google Scholar]
- Ciccia, A.; McDonald, N.; West, S.C. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 2008, 77, 259–287. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Ulrich, C.M.; Potter, J.D. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol. Biomarkers Prev. 2002, 11, 1513–1530. [Google Scholar] [PubMed]
- Cook, J.S. Photoreactivation in animal cells. Photophysiology 1970, 5, 191–233. [Google Scholar] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington DC, WA, USA, 2006; p. 1118. [Google Scholar]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968, 218, 652–656. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Lee, C.-H.; Seo, Y.-S. Lagging Strand Synthesis and Genomic Stability. In DNA Repair: On the Pathways to Fixing DNA Damage and Errors; Storici, F., Ed.; InTech: Rijeka, Croatia, 2011; pp. 1–14. [Google Scholar]
- Manandhar, M.; Boulware, K.S.; Wood, R.D. The ERCC1 and ERCC4 (XPF) genes and gene products. Gene 2015, 569, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Liu, X.; Li, L.; Legerski, R. Double-strand breaks induce homologous recombinational repair of interstrand cross-links via cooperation of MSH2, ERCC1-XPF, REV3, and the Fanconi anemia pathway. DNA Repair (Amst) 2007, 6, 1670–1678. [Google Scholar] [CrossRef] [Green Version]
- Gillet, L.C.; Scharer, O.D. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006, 106, 253–276. [Google Scholar] [CrossRef]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Xu, Y.; Qiu, L.X.; Li, J.; Zhou, X.Y.; Sun, M.H.; Wang, J.C.; Yang, Y.J.; Jin, L.; Wei, Q.Y.; et al. Polymorphisms in ERCC1 and XPF genes and risk of gastric cancer in an eastern Chinese population. PLoS One 2012, 7, e49308. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Alkyltransferase-like Proteins: Brokers Dealing with Alkylated DNA Bases. Mol. Cell 2012, 47, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.; Melton, D.W. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010, 30, 3223–3232. [Google Scholar] [PubMed]
- Min, J.H.; Pavletich, N.P. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 2007, 449, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Ng, J.M.Y.; Masutani, C.; Iwai, S.; van der Spek, P.J.; Eker, A.P.M.; Hanaoka, F.; Bootsma, D.; Hoeijmakers, J.H.J. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 1998, 2, 223–232. [Google Scholar] [CrossRef]
- Volkert, M.R.; Landini, P. Transcriptional responses to DNA damage. Curr. Opin. Microbiol. 2001, 4, 178–185. [Google Scholar] [CrossRef]
- Ellenberger, T. An arresting development in transcription. Mol. Cell 2012, 46, 3–4. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Hanawalt, P.C. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002, 21, 8949–8956. [Google Scholar] [CrossRef] [Green Version]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenet. Chromatin 2012, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.J. Blinded by the UV light: How the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair (Amst) 2013, 12, 656–671. [Google Scholar] [CrossRef] [Green Version]
- Gregersen, L.H.; Svejstrup, J.Q. The Cellular Response to Transcription-Blocking DNA Damage. Trends Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signaling 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Faridounnia, M.; Kovacic, L.; Kaptein, R.; Boelens, R.; Folkers, G.E. Single-stranded DNA Binding by the Helix-Hairpin-Helix Domain of XPF Protein Contributes to the Substrate Specificity of the ERCC1-XPF Protein Complex. J. Biol. Chem. 2017, 292, 2842–2853. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Coin, F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair 2011, 10, 714–721. [Google Scholar] [CrossRef]
- Matsunaga, T.; Park, C.H.; Bessho, T.; Mu, D.; Sancar, A. Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nucleases. J. Biol. Chem. 1996, 271, 11047–11050. [Google Scholar] [CrossRef]
- Fisher, L.A.; Bessho, M.; Wakasugi, M.; Matsunaga, T.; Bessho, T. Role of interaction of XPF with RPA in nucleotide excision repair. J. Mol. Biol. 2011, 413, 337–346. [Google Scholar] [CrossRef]
- Park, C.H.; Sancar, A. Formation of a ternary complex by human XPA, ERCC1, and ERCC4(XPF) excision repair proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 5017–5021. [Google Scholar] [CrossRef]
- Saijo, M.; Kuraoka, I.; Masutani, C.; Hanaoka, F.; Tanaka, K. Sequential binding of DNA repair proteins RPA and ERCC1 to XPA in vitro. Nucleic Acids Res. 1996, 24, 4719–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripsianes, K.; Folkers, G.E.; Zheng, C.; Das, D.; Grinstead, J.S.; Kaptein, R.; Boelens, R. Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res. 2007, 35, 5789–5798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsodikov, O.V.; Ivanov, D.; Orelli, B.; Staresincic, L.; Shoshani, I.; Oberman, R.; Scharer, O.D.; Wagner, G.; Ellenberger, T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007, 26, 4768–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orelli, B.; McClendon, T.B.; Tsodikov, O.V.; Ellenberger, T.; Niedernhofer, L.J.; Scharer, O.D. The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. J. Biol. Chem. 2010, 285, 3705–3712. [Google Scholar] [CrossRef] [PubMed]
- Staresincic, L.; Fagbemi, A.F.; Enzlin, J.H.; Gourdin, A.M.; Wijgers, N.; Dunand-Sauthier, I.; Giglia-Mari, G.; Clarkson, S.G.; Vermeulen, W.; Scharer, O.D. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009, 28, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.F.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.R.; Prakash, L. Incision and postincision steps of pyrimidine dimer removal in excision-defective mutants of Saccharomyces cerevisiae. J. Bacteriol. 1981, 148, 618–623. [Google Scholar]
- Davies, A.A.; Friedberg, E.C.; Tomkinson, A.E.; Wood, R.D.; West, S.C. Role of the Rad1 and Rad10 proteins in nucleotide excision repair and recombination. J. Biol. Chem. 1995, 270, 24638–24641. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Bardwell, L.; Tomkinson, A.E.; Friedberg, E.C. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 1994, 265, 2082–2085. [Google Scholar] [CrossRef]
- Brookman, K.W.; Lamerdin, J.E.; Thelen, M.P.; Hwang, M.; Reardon, J.T.; Sancar, A.; Zhou, Z.Q.; Walter, C.A.; Parris, C.N.; Thompson, L.H. ERCC4 (XPF) encodes a human nucleotide excision repair protein with eukaryotic recombination homologs. Mol. Cell. Biol. 1996, 16, 6553–6562. [Google Scholar] [CrossRef]
- Van Duin, M.; de Wit, J.; Odijk, H.; Westerveld, A.; Yasui, A.; Koken, M.H.; Hoeijmakers, J.H.; Bootsma, D. Molecular characterization of the human excision repair gene ERCC-1: cDNA cloning and amino acid homology with the yeast DNA repair gene RAD10. Cell 1986, 44, 913–923. [Google Scholar] [CrossRef]
- De Laat, W.L.; Appeldoorn, E.; Jaspers, N.G.; Hoeijmakers, J.H. DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 1998, 273, 7835–7842. [Google Scholar] [CrossRef] [PubMed]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, P.J.; Spanswick, V.J.; Hartley, J.A. Repair of DNA interstrand crosslinks: Molecular mechanisms and clinical relevance. Lancet Oncol. 2001, 2, 483–490. [Google Scholar] [CrossRef]
- Goodarzi, A.; Jeggo, P. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2012, 82, 1–45. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Ahmad, A.; Robinson, A.R.; Duensing, A.; van Drunen, E.; Beverloo, H.B.; Weisberg, D.B.; Hasty, P.; Hoeijmakers, J.H.; Niedernhofer, L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008, 28, 5082–5092. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, M.; Watanabe, S.; Johnson, A.; Moazed, D.; Peterson, C.L. Recombinational Repair within Heterochromatin Requires ATP-Dependent Chromatin Remodeling. Cell 2009, 138, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaskelioff, M.; Van Komen, S.; Krebs, J.E.; Sung, P.; Peterson, C.L. Rad54p is a chromatin remodeling enzyme required for heteroduplex DNA joint formation with chromatin. J. Biol. Chem. 2003, 278, 9212–9218. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; de Massy, B. Regulating double-stranded DNA break repair towards crossover or non-crossover during mammalian meiosis. Chromosome Res. 2007, 15, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrentino, M.-E.; Borde, V.R. The spatial regulation of meiotic recombination hotspots: Are all DSB hotspots crossover hotspots? Exp. Cell Res. 2012, 318, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.D.; Li, X.; Rolfsmeier, M.; Zhang, X.P. Rad54: The Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006, 34, 4115–4125. [Google Scholar] [CrossRef]
- Wyatt, H.D.; Laister, R.C.; Martin, S.R.; Arrowsmith, C.H.; West, S.C. The SMX DNA Repair Tri-nuclease. Mol. Cell 2017, 65, 848–860. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Pezza, R.J.; Mazina, O.M.; Voloshin, O.N.; Camerini-Otero, R.D.; Mazin, A.V. The resistance of DMC1 D-loops to dissociation may account for the DMC1 requirement in meiosis. Nat. Struct. Mol. Biol. 2010, 18, 56–60. [Google Scholar] [CrossRef]
- Al-Minawi, A.Z.; Saleh-Gohari, N.; Helleday, T. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 2008, 36, 1–9. [Google Scholar] [CrossRef]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.K.; Haber, J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Yu, Y.; Mahaney, B.L.; Cai, B.; Ye, R.; Phipps, B.M.; Rambo, R.P.; Hura, G.L.; Pelikan, M.; So, S.; et al. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem. 2010, 285, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Yu, Y.; Radhakrishnan, S.K.; Chokshi, C.; Tsai, M.S.; Matsumoto, Y.; Kuzdovich, M.; Remesh, S.G.; Fang, S.; Tomkinson, A.E.; et al. An Intrinsically Disordered APLF Links Ku, DNA-PKcs, and XRCC4-DNA Ligase IV in an Extended Flexible Non-homologous End Joining Complex. J. Biol. Chem. 2016, 291, 26987–27006. [Google Scholar] [CrossRef] [PubMed]
- Corbeski, I.; Dolinar, K.; Wienk, H.; Boelens, R.; van Ingen, H. DNA repair factor APLF acts as a H2A-H2B histone chaperone through binding its DNA interaction surface. Nucleic Acids Res. 2018, 46, 7138–7152. [Google Scholar] [CrossRef]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef]
- Wu, H.; Roks, A.J. Genomic instability and vascular aging: A focus on nucleotide excision repair. Trends Cardiovasc. Med. 2014, 24, 61–68. [Google Scholar] [CrossRef]
- McDaniel, L.D.; Schultz, R.A. XPF/ERCC4 and ERCC1: Their products and biological roles. In Molecular Mechanisms of Xeroderma Pigmentosum; Ahmad, S.I., Hanaoka, F., Eds.; Springer: New York, NY, USA, 2008; Volume 637, pp. 65–82. [Google Scholar]
- Lehmann, J.; Seebode, C.; Smolorz, S.; Schubert, S.; Emmert, S. XPF knockout via CRISPR/Cas9 reveals that ERCC1 is retained in the cytoplasm without its heterodimer partner XPF. Cell. Mol. Life Sci. 2017, 74, 2081–2094. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVey, M. RPA puts the brakes on MMEJ. Nat. Struct. Mol. Biol. 2014, 21, 348–349. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair (Amst) 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Nakajima, S.; Hsieh, C.L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage response of XRCC1 at sites of DNA single strand breaks is regulated by phosphorylation and ubiquitylation after degradation of poly(ADP-ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Takahata, C.; Masuda, Y.; Takedachi, A.; Tanaka, K.; Iwai, S.; Kuraoka, I. Repair synthesis step involving ERCC1-XPF participates in DNA repair of the Top1-DNA damage complex. Carcinogenesis 2015, 36, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.L.; Gottesman, M.E.; Gautier, J. The differences between ICL repair during and outside of S phase. Trends Biochem. Sci. 2013, 38, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, L. DNA crosslinking damage and cancer–a tale of friend and foe. Transl. Cancer. Res. 2013, 2, 144–154. [Google Scholar] [CrossRef]
- Czerwinska, J.; Nowak, M.; Wojtczak, P.; Dziuban-Lech, D.; Ciesla, J.M.; Kolata, D.; Gajewska, B.; Baranczyk-Kuzma, A.; Robinson, A.R.; Shane, H.L.; et al. ERCC1-deficient cells and mice are hypersensitive to lipid peroxidation. Free Radic. Biol. Med. 2018, 124, 79–96. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Kim, J.M.; Shiotani, B.; Yang, K.L.; Zou, L.; D’Andrea, A.D. The FANCM/FAAP24 Complex Is Required for the DNA Interstrand Crosslink-Induced Checkpoint Response. Mol. Cell 2010, 39, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lambert, M.W. The Fanconi anemia protein, FANCG, binds to the ERCC1-XPF endonuclease via its tetratricopeptide repeats and the central domain of ERCC1. Biochemistry 2010, 49, 5560–5569. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. How the Fanconi Anemia Pathway Guards the Genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walden, H.; Deans, A.J. The Fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Ren, K. Fanconi anemia proteins, DNA interstrand crosslink repair pathways, and cancer therapy. Curr. Cancer Drug Targets 2009, 9, 101–117. [Google Scholar] [CrossRef]
- Abdullah, U.B.; McGouran, J.F.; Brolih, S.; Ptchelkine, D.; El-Sagheer, A.H.; Brown, T.; McHugh, P.J. RPA activates the XPF-ERCC1 endonuclease to initiate processing of DNA interstrand crosslinks. EMBO J. 2017, 36, 2047–2060. [Google Scholar] [CrossRef]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef]
- Miles, J.A.; Frost, M.G.; Carroll, E.; Rowe, M.L.; Howard, M.J.; Sidhu, A.; Chaugule, V.K.; Alpi, A.F.; Walden, H. The Fanconi Anemia DNA Repair Pathway Is Regulated by an Interaction between Ubiquitin and the E2-like Fold Domain of FANCL. J. Biol. Chem. 2015, 290, 20995–21006. [Google Scholar] [CrossRef]
- Klein Douwel, D.; Boonen, R.A.; Long, D.T.; Szypowska, A.A.; Raschle, M.; Walter, J.C.; Knipscheer, P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell 2014, 54, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Adair, G.M.; Rolig, R.L.; Moore-Faver, D.; Zabelshansky, M.; Wilson, J.H.; Nairn, R.S. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000, 19, 5552–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagwat, N.; Olsen, A.L.; Wang, A.T.; Hanada, K.; Stuckert, P.; Kanaar, R.; D’Andrea, A.; Niedernhofer, L.J.; McHugh, P.J. XPF-ERCC1 Participates in the Fanconi Anemia Pathway of Cross-Link Repair. Mol. Cell. Biol. 2009, 29, 6427–6437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef]
- Klein Douwel, D.; Hoogenboom, W.S.; Boonen, R.A.; Knipscheer, P. Recruitment and positioning determine the specific role of the XPF-ERCC1 endonuclease in interstrand crosslink repair. EMBO J. 2017, 36, 2034–2046. [Google Scholar] [CrossRef]
- Seol, J.H.; Holland, C.; Li, X.; Kim, C.; Li, F.; Medina-Rivera, M.; Eichmiller, R.; Gallardo, I.F.; Finkelstein, I.J.; Hasty, P.; et al. Distinct roles of XPF-ERCC1 and Rad1-Rad10-Saw1 in replication-coupled and uncoupled inter-strand crosslink repair. Nat. Commun. 2018, 9, 2025. [Google Scholar] [CrossRef]
- Vuono, E.A.; Mukherjee, A.; Vierra, D.A.; Adroved, M.M.; Hodson, C.; Deans, A.J.; Howlett, N.G. The PTEN phosphatase functions cooperatively with the Fanconi anemia proteins in DNA crosslink repair. Sci. Rep. 2016, 6, 36439. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.M.; Hain, K.; Déclais, A.-C.; Gardiner, M.; Toh, G.W.; Sanchez-Pulido, L.; Heuckmann, J.M.; Toth, R.; Macartney, T.; Eppink, B.; et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 2009, 35, 116–127. [Google Scholar] [CrossRef]
- Hodskinson, M.R.; Silhan, J.; Crossan, G.P.; Garaycoechea, J.I.; Mukherjee, S.; Johnson, C.M.; Scharer, O.D.; Patel, K.J. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell 2014, 54, 472–484. [Google Scholar] [CrossRef]
- McNeil, E.M.; Melton, D.W. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012, 40, 9990–10004. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschle, M.; Knipscheer, P.; Enoiu, M.; Angelov, T.; Sun, J.; Griffith, J.D.; Ellenberger, T.E.; Scharer, O.D.; Walter, J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008, 134, 969–980. [Google Scholar] [CrossRef]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef]
- Van Cuijk, L.; Vermeulen, W.; Marteijn, J.A. Ubiquitin at work: The ubiquitous regulation of the damage recognition step of NER. Exp. Cell Res. 2014, 329, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chitale, S.; Richly, H. Timing of DNA lesion recognition: Ubiquitin signaling in the NER pathway. Cell Cycle 2017, 16, 163–171. [Google Scholar] [CrossRef]
- Zhang, L.; Gong, F. The emerging role of deubiquitination in nucleotide excision repair. DNA Repair (Amst) 2016, 44, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cuijk, L.; van Belle, G.J.; Turkyilmaz, Y.; Poulsen, S.L.; Janssens, R.C.; Theil, A.F.; Sabatella, M.; Lans, H.; Mailand, N.; Houtsmuller, A.B.; et al. SUMO and ubiquitin-dependent XPC exchange drives nucleotide excision repair. Nat. Commun. 2015, 6, 7499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Ritchie, A.M.; Melton, D.W. Disruption of DNA repair in cancer cells by ubiquitination of a destabilising dimerization domain of nucleotide excision repair protein ERCC1. Oncotarget 2017, 8, 55246–55264. [Google Scholar] [CrossRef] [PubMed]
- Perez-Oliva, A.B.; Lachaud, C.; Szyniarowski, P.; Munoz, I.; Macartney, T.; Hickson, I.; Rouse, J.; Alessi, D.R. USP45 deubiquitylase controls ERCC1-XPF endonuclease-mediated DNA damage responses. EMBO J. 2015, 34, 326–343. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.L.; Gottesman, M.E.; Gautier, J. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 2012, 47, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Hayashi, T.; Rabeya, R.M.; Nakajima, S.; Kanno, S.; Takao, M.; Matsunaga, T.; Yoshino, M.; Ichikawa, M.; Riele, H.T.; et al. Functional and physical interactions between ERCC1 and MSH2 complexes for resistance to cis-diamminedichloroplatinum(II) in mammalian cells. DNA Repair 2004, 3, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Claij, N.; Te Riele, H. Methylation tolerance in mismatch repair proficient cells with low MSH2 protein level. Oncogene 2002, 21, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Kothandapani, A.; Sawant, A.; Dangeti, V.S.; Sobol, R.W.; Patrick, S.M. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res. 2013, 41, 7332–7343. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, P.; Tishkoff, D.X.; Filosi, N.; Dasgupta, R.; Kolodner, R.D. Physical interaction between components of DNA mismatch repair and nucleotide excision repair. Proc. Natl. Acad. Sci. USA 1998, 95, 14278–14283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, P.; Blanco, R.; Flores, J.M.; Blasco, M.A. XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat. Genet. 2005, 37, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Niedernhofer, L.; Kuster, B.; Mann, M.; Hoeijmakers, J.H.; de Lange, T. ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 2003, 12, 1489–1498. [Google Scholar] [CrossRef]
- Svendsen, J.M.; Smogorzewska, A.; Sowa, M.E.; O’Connell, B.C.; Gygi, S.P.; Elledge, S.J.; Harper, J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009, 138, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Yin, J.; Horvath, K.; Sarkar, J.; Chen, Y.; Wu, J.; Wan, K.; Lu, J.; Gu, P.; Yu, E.Y.; et al. SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep. 2013, 4, 861–869. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zacal, N.J.; Rainbow, A.J.; Zhu, X.D. XPF with mutations in its conserved nuclease domain is defective in DNA repair but functions in TRF2-mediated telomere shortening. DNA Repair 2007, 6, 157–166. [Google Scholar] [CrossRef]
- Naim, V.; Wilhelm, T.; Debatisse, M.; Rosselli, F. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 2013, 15, 1008–1015. [Google Scholar] [CrossRef]
- Kamileri, I.; Karakasilioti, I.; Sideri, A.; Kosteas, T.; Tatarakis, A.; Talianidis, I.; Garinis, G.A. Defective transcription initiation causes postnatal growth failure in a mouse model of nucleotide excision repair (NER) progeria. Proc. Natl. Acad. Sci. USA 2012, 109, 2995–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzinikolaou, G.; Apostolou, Z.; Aid-Pavlidis, T.; Ioannidou, A.; Karakasilioti, I.; Papadopoulos, G.L.; Aivaliotis, M.; Tsekrekou, M.; Strouboulis, J.; Kosteas, T.; et al. ERCC1-XPF cooperates with CTCF and cohesin to facilitate the developmental silencing of imprinted genes. Nat. Cell Biol. 2017, 19, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Velez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell. Mol. Life Sci. 2009, 66, 994–1009. [Google Scholar] [CrossRef] [PubMed]
- Rass, U.; Ahel, I.; West, S.C. Defective DNA repair and neurodegenerative disease. Cell 2007, 130, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Nishigori, C.; Yagi, T.; Imamura, S.; Takebe, H. Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum. Mol. Genet. 1998, 7, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Gurkar, A.U.; Niedernhofer, L.J. Comparison of mice with accelerated aging caused by distinct mechanisms. Exp. Gerontol. 2015, 68, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Sijbers, A.M.; van Voorst Vader, P.C.; Snoek, J.W.; Raams, A.; Jaspers, N.G.; Kleijer, W.J. Homozygous R788W point mutation in the XPF gene of a patient with xeroderma pigmentosum and late-onset neurologic disease. J. Invest. Dermatol. 1998, 110, 832–836. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Imoto, K.; Boyle, J.; Oh, K.; Khan, S.; Ueda, T.; Nadem, C.; Slor, H.; Orgal, S.; Gadoth, N.; Busch, D.; et al. Patients with defects in the interacting nucleotide excision repair proteins ERCC1 or XPF show xeroderma pigmentosum with late onset severe neurological degeneration. In Proceedings of the 68th Annual Meeting of the Society for Investigative Dermatology, Los Angeles, California, CA, USA, 9–12 May 2007; Abstract 547. p. S92. [Google Scholar] [CrossRef] [Green Version]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. The many faces of Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15273–15274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brace, L.E.; Vose, S.C.; Vargas, D.F.; Zhao, S.; Wang, X.P.; Mitchell, J.R. Lifespan extension by dietary intervention in a mouse model of Cockayne syndrome uncouples early postnatal development from segmental progeria. Aging Cell 2013, 12, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 2003, 85, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- De Waard, H.; de Wit, J.; Andressoo, J.O.; van Oostrom, C.T.; Riis, B.; Weimann, A.; Poulsen, H.E.; van Steeg, H.; Hoeijmakers, J.H.; van der Horst, G.T. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol. Cell. Biol. 2004, 24, 7941–7948. [Google Scholar] [CrossRef]
- Trego, K.S.; Groesser, T.; Davalos, A.R.; Parplys, A.C.; Zhao, W.; Nelson, M.R.; Hlaing, A.; Shih, B.; Rydberg, B.; Pluth, J.M.; et al. Non-catalytic Roles for XPG with BRCA1 and BRCA2 in Homologous Recombination and Genome Stability. Mol. Cell 2016, 61, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Barnhoorn, S.; Uittenboogaard, L.M.; Jaarsma, D.; Vermeij, W.P.; Tresini, M.; Weymaere, M.; Menoni, H.; Brandt, R.M.; de Waard, M.C.; Botter, S.M.; et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 2014, 10, e1004686. [Google Scholar] [CrossRef]
- Sabatella, M.; Theil, A.F.; Ribeiro-Silva, C.; Slyskova, J.; Thijssen, K.; Voskamp, C.; Lans, H.; Vermeulen, W. Repair protein persistence at DNA lesions characterizes XPF defect with Cockayne syndrome features. Nucleic Acids Res. 2018, 46, 9563–9577. [Google Scholar] [CrossRef]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, N.G.; Raams, A.; Silengo, M.C.; Wijgers, N.; Niedernhofer, L.J.; Robinson, A.R.; Giglia-Mari, G.; Hoogstraten, D.; Kleijer, W.J.; Hoeijmakers, J.H.; et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 2007, 80, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Andressoo, J.O.; Hoeijmakers, J.H. Transcription-coupled repair and premature ageing. Mutat. Res. 2005, 577, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Enzlin, J.H.; Bhagwat, N.R.; Wijgers, N.; Raams, A.; Appledoorn, E.; Theil, A.F.; JH, J.H.; Vermeulen, W.; NG, J.J.; et al. Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet. 2010, 6, e1000871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faridounnia, M.; Wienk, H.; Kovacic, L.; Folkers, G.E.; Jaspers, N.G.J.; Kaptein, R.; Hoeijmakers, J.H.J.; Boelens, R. The Cerebro-oculo-facio-skeletal Syndrome Point Mutation F231L in the ERCC1 DNA Repair Protein Causes Dissociation of the ERCC1-XPF Complex. J. Biol. Chem. 2015, 290, 20541–20555. [Google Scholar] [CrossRef] [PubMed]
- Bessho, T.; Sancar, A.; Thompson, L.H.; Thelen, M.P. Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J. Biol. Chem. 1997, 272, 3833–3837. [Google Scholar] [CrossRef] [PubMed]
- Moggs, J.G.; Yarema, K.J.; Essigmann, J.M.; Wood, R.D. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J. Biol. Chem. 1996, 271, 7177–7186. [Google Scholar] [CrossRef]
- Osorio, A.; Bogliolo, M.; Fernandez, V.; Barroso, A.; de la Hoya, M.; Caldes, T.; Lasa, A.; Ramon y Cajal, T.; Santamarina, M.; Vega, A.; et al. Evaluation of rare variants in the new fanconi anemia gene ERCC4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum. Mutat. 2013, 34, 1615–1618. [Google Scholar] [CrossRef]
- Eisen, J.A.; Hanawalt, P.C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 1999, 435, 171–213. [Google Scholar] [CrossRef] [Green Version]
- Sgouros, J.; Gaillard, P.H.L.; Wood, R.D. A relationship between a DNA-repair/recombination nuclease family and archaeal helicases. Trends Biochem. Sci. 1999, 24, 95–97. [Google Scholar] [CrossRef]
- Singh, S.; Folkers, G.E.; Bonvin, A.M.J.J.; Boelens, R.; Wechselberger, R.; Niztayev, A.; Kaptein, R. Solution structure and DNA-binding properties of the C-terminal domain of UvrC from E.coli. EMBO J. 2002, 21, 6257–6266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L.; Park, J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falquet, L.; Bordoli, L.; Ioannidis, V.; Pagni, M.; Jongeneel, C.V. Swiss EMBnet node web server. Nucleic Acids Res. 2003, 31, 3782–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, A.J.; Serpell, L.C.; Ponting, C.P. The helix-hairpin-helix DNA-binding motif: A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996, 24, 2488–2497. [Google Scholar] [CrossRef] [PubMed]
- Wienk, H.; Slootweg, J.C.; Speerstra, S.; Kaptein, R.; Boelens, R.; Folkers, G.E. The Fanconi anemia associated protein FAAP24 uses two substrate specific binding surfaces for DNA recognition. Nucleic Acids Res. 2013, 41, 6739–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Han, X.; Wu, F.; Leung, J.W.; Lowery, M.G.; Do, H.; Chen, J.; Shi, C.; Tian, C.; Li, L.; et al. Structure analysis of FAAP24 reveals single-stranded DNA-binding activity and domain functions in DNA damage response. Cell Res. 2013, 23, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Enzlin, J.H.; Scharer, O.D. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002, 21, 2045–2053. [Google Scholar] [CrossRef] [Green Version]
- Youds, J.L.; O’Neil, N.J.; Rose, A.M. Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 2006, 173, 697–708. [Google Scholar] [CrossRef]
- Tripsianes, K.; Folkers, G.; Ab, E.; Das, D.; Odijk, H.; Jaspers, N.G.; Hoeijmakers, J.H.; Kaptein, R.; Boelens, R. The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure 2005, 13, 1849–1858. [Google Scholar] [CrossRef]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; West, S.C. MUS81-EME2 promotes replication fork restart. Cell Rep 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, T.L.; Tao, Y.; Wang, F.; Tong, L.; Ding, J.P. Structural insights into the functions of the FANCM-FAAP24 complex in DNA repair. Nucleic Acids Res. 2013, 41, 10573–10583. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kee, Y.; Gurtan, A.; D’Andrea, A.D. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 2008, 111, 5215–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcas, A.; Fernandez-Capetillo, O.; Cases, I.; Rojas, A.M. Emergence and evolutionary analysis of the human DDR network: Implications in comparative genomics and downstream analyses. Mol. Biol. Evol. 2014, 31, 940–961. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.H.L.; Wood, R.D. Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res. 2001, 29, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsodikov, O.V.; Enzlin, J.H.; Scharer, O.D.; Ellenberger, T. Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc. Natl. Acad. Sci. USA 2005, 102, 11236–11241. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Xue, Y.T.; Ling, C.; Takata, M.; Wang, W.D.; Keck, J.L. Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc. Natl. Acad. Sci. USA 2012, 109, 4437–4442. [Google Scholar] [CrossRef] [Green Version]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Mosedale, G.; Niedzwiedz, W.; Alpi, A.; Perrina, F.; Pereira-Leal, J.B.; Johnson, M.; Langevin, F.; Pace, P.; Patel, K.J. The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat. Struct. Mol. Biol. 2005, 12, 763–771. [Google Scholar] [CrossRef]
- Rosado, I.V.; Niedzwiedz, W.; Alpi, A.F.; Patel, K.J. The Walker B motif in avian FANCM is required to limit sister chromatid exchanges but is dispensable for DNA crosslink repair. Nucleic Acids Res. 2009, 37, 4360–4370. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Li, Y.; Guo, R.; Ling, C.; Wang, W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Genet. 2008, 17, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Yousefzadeh, M.J.; Faridounnia, M.; Chong, J.X.; Hisama, F.M.; Hudgins, L.; Mercado, G.; Wade, E.A.; Barghouthy, A.S.; Lee, L.; et al. ERCC4 variants identified in a cohort of patients with segmental progeroid syndromes. Hum. Mutat. 2018, 39, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Walker, D.R.; Koonin, E.V. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999, 27, 1223–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Komori, K.; Ishino, Y.; Morikawa, K. X-ray and biochemical anatomy of an archaeal XPF/Rad1/Mus81 family nuclease: Similarity between its endonuclease domain and restriction enzymes. Structure 2003, 11, 445–457. [Google Scholar] [CrossRef]
- Pingoud, A.; Jeltsch, A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001, 29, 3705–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Elledge, S.J.; Peterson, C.A.; Bales, E.S.; Legerski, R.J. Specific Association between the Human DNA-Repair Proteins Xpa and Ercc1. Proc. Natl. Acad. Sci. USA. 1994, 91, 5012–5016. [Google Scholar] [CrossRef]
- Shao, X.G.; Grishin, N.V. Common fold in helix-hairpin-helix proteins. Nucleic Acids Res. 2000, 28, 2643–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.; Murray-Rust, J.; Lally, J.; Rudolf, J.; Fadden, A.; Knowles, P.P.; White, M.F.; McDonald, N.Q. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 2005, 24, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Folkers, G.E.; van Dijk, M.; Jaspers, N.G.; Hoeijmakers, J.H.; Kaptein, R.; Boelens, R. The structure of the XPF-ssDNA complex underscores the distinct roles of the XPF and ERCC1 helix- hairpin-helix domains in ss/ds DNA recognition. Structure 2012, 20, 667–675. [Google Scholar] [CrossRef]
- De Laat, W.L.; Sijbers, A.M.; Odijk, H.; Jaspers, N.G.; Hoeijmakers, J.H. Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res. 1998, 26, 4146–4152. [Google Scholar] [CrossRef] [Green Version]
- Jordheim, L.P.; Barakat, K.H.; Heinrich-Balard, L.; Matera, E.L.; Cros-Perrial, E.; Bouledrak, K.; El Sabeh, R.; Perez-Pineiro, R.; Wishart, D.S.; Cohen, R.; et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol. Pharmacol. 2013, 84, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, F.; Barakat, K.H.; Tuszynski, J.A. Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease. Int. J. Mol. Sci. 2018, 19, 1328. [Google Scholar] [CrossRef]
- Liccardi, G.; Hartley, J.A.; Hochhauser, D. Importance of EGFR/ERCC1 interaction following radiation-induced DNA damage. Clin. Cancer Res. 2014, 20, 3496–3506. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, M.B.; Shen, J.P.; Gross, A.M.; Huang, J.K.; Ideker, T.; Fanta, P. ERCC1 and TS Expression as Prognostic and Predictive Biomarkers in Metastatic Colon Cancer. PLoS One 2015, 10, e0126898. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fang, Y.J.; Li, F.; Ou, Q.J.; Chen, G.; Ma, G. ERCC1, defective mismatch repair status as predictive biomarkers of survival for stage III colon cancer patients receiving oxaliplatin-based adjuvant chemotherapy. Br. J. Cancer 2013, 108, 1238–1244. [Google Scholar] [CrossRef] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. https://doi.org/10.3390/molecules23123205
Faridounnia M, Folkers GE, Boelens R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules. 2018; 23(12):3205. https://doi.org/10.3390/molecules23123205
Chicago/Turabian StyleFaridounnia, Maryam, Gert E. Folkers, and Rolf Boelens. 2018. "Function and Interactions of ERCC1-XPF in DNA Damage Response" Molecules 23, no. 12: 3205. https://doi.org/10.3390/molecules23123205
APA StyleFaridounnia, M., Folkers, G. E., & Boelens, R. (2018). Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules, 23(12), 3205. https://doi.org/10.3390/molecules23123205