Microhydration and the Enhanced Acidity of Free Radicals

Abstract
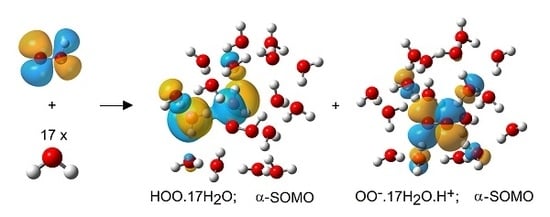
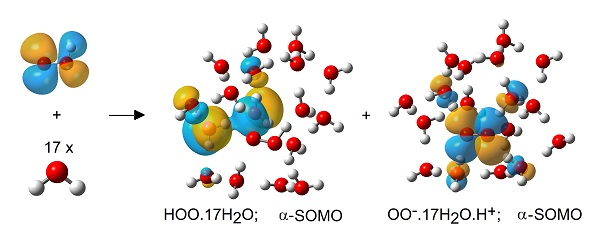
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
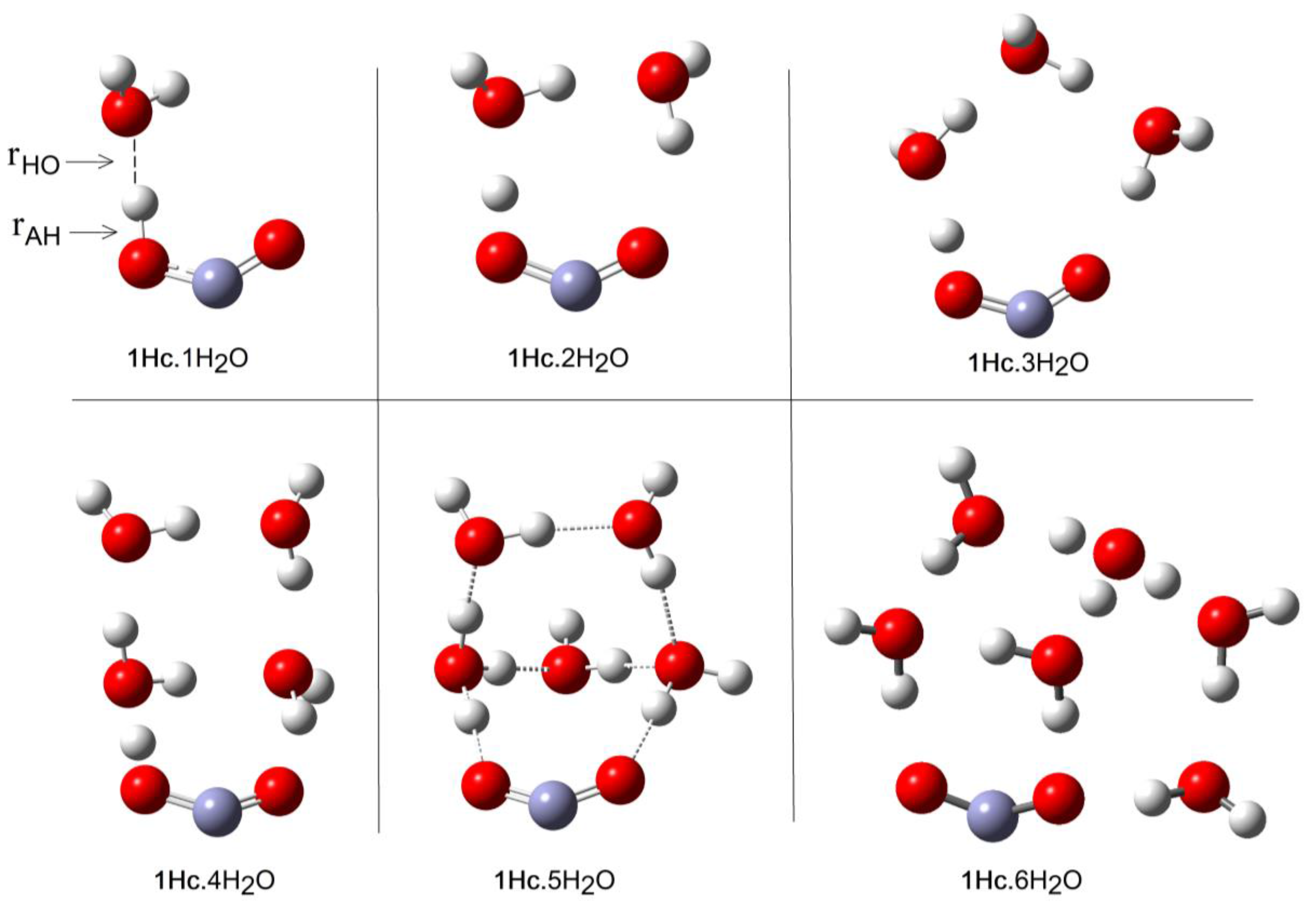
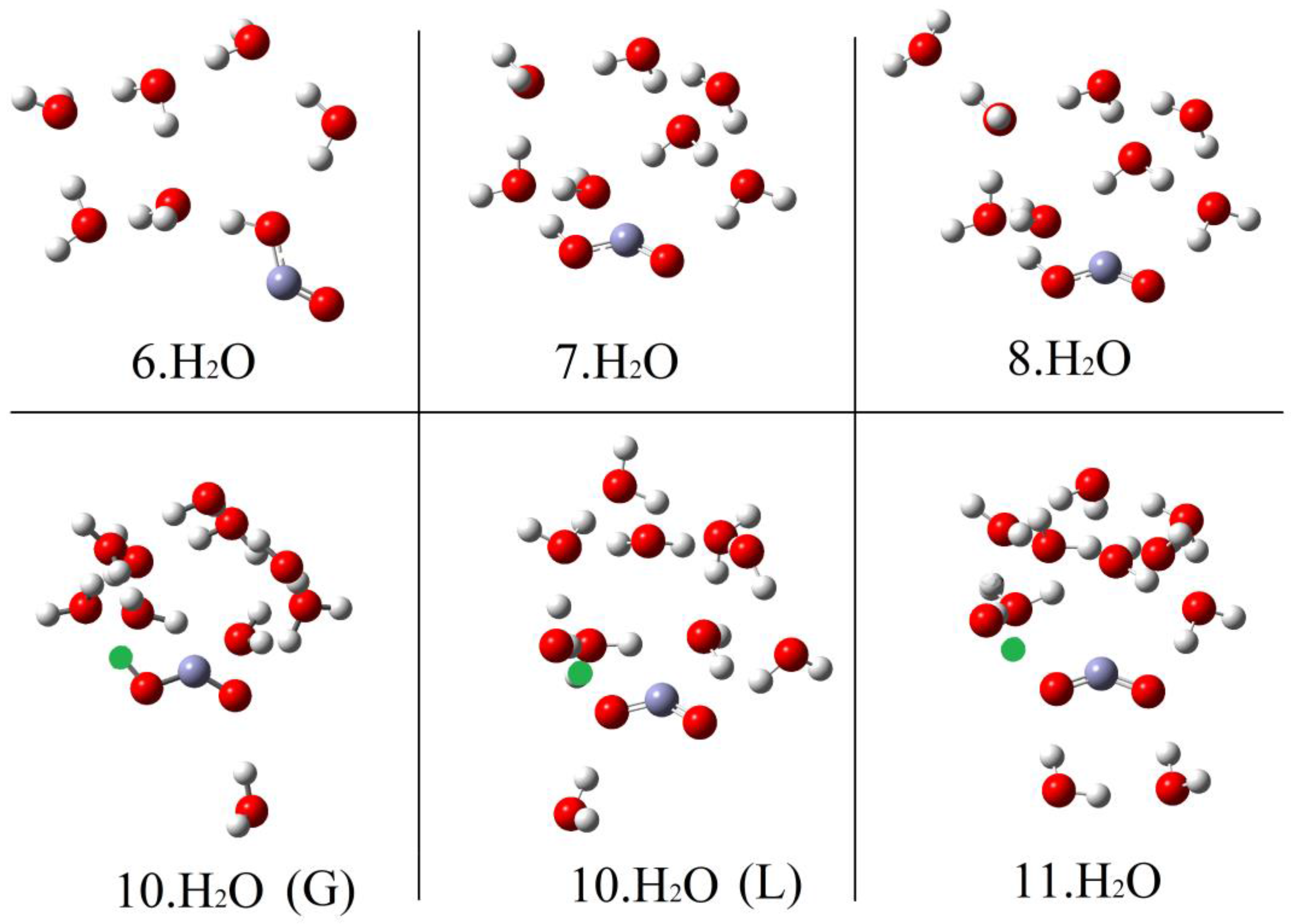
2.1. Microhydration of Carboxyl Radicals 1Hc and 1Ht
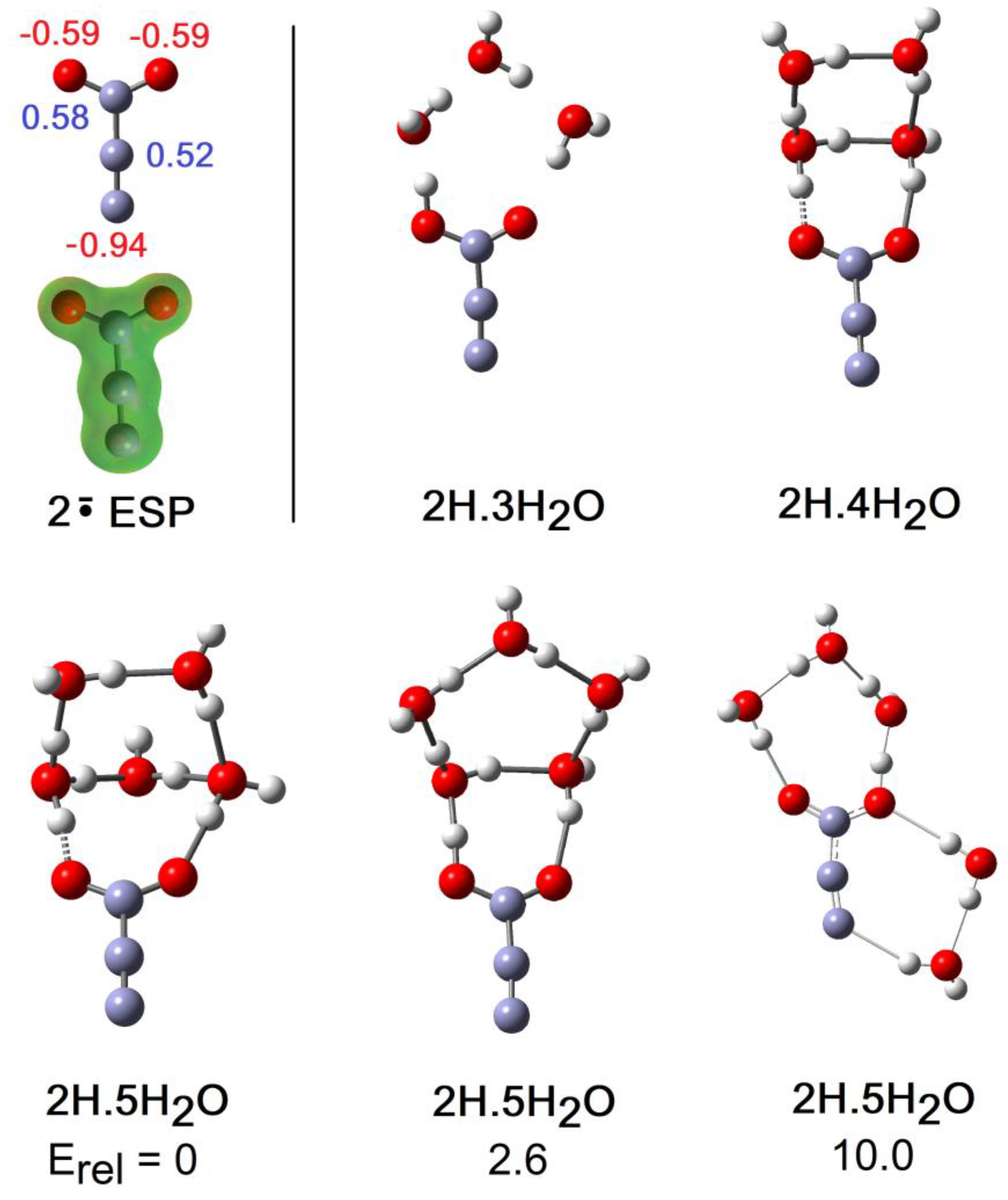
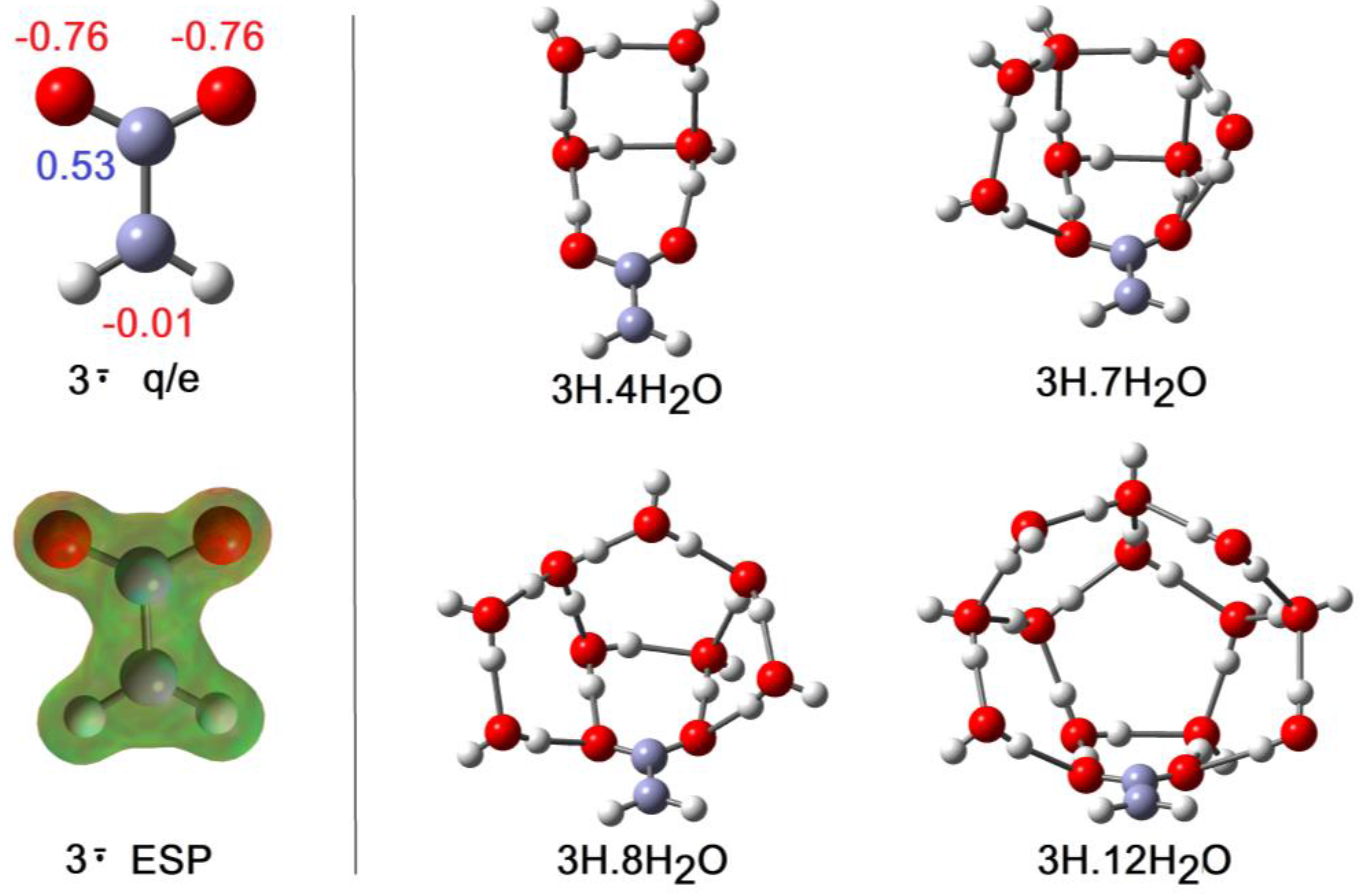
2.2. Microhydration of Carboxy-ethynyl 2H and Carboxy-methyl 3H Radicals
2.3. Microhydration of Hydroperoxyl Radicals 5H
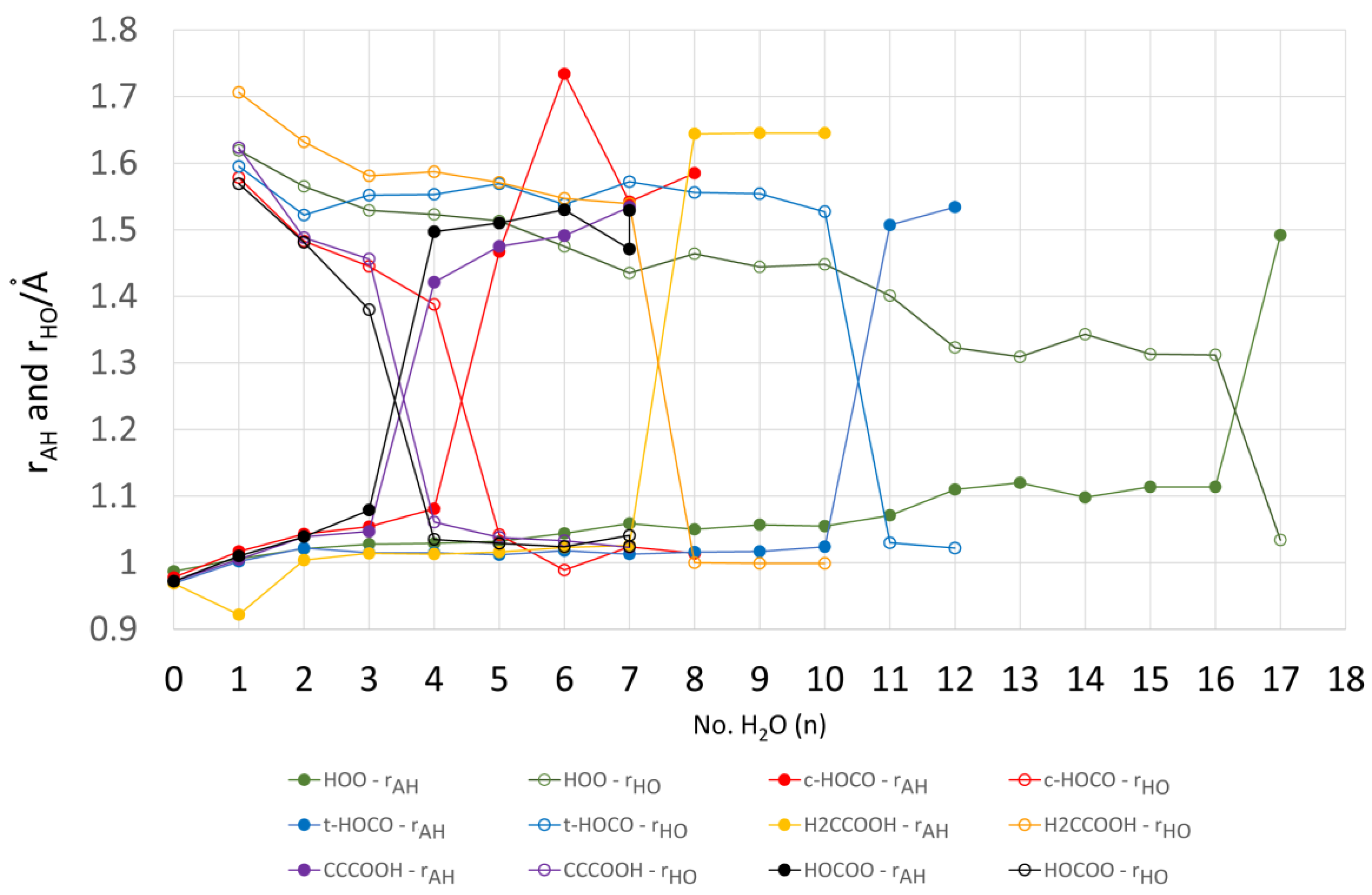
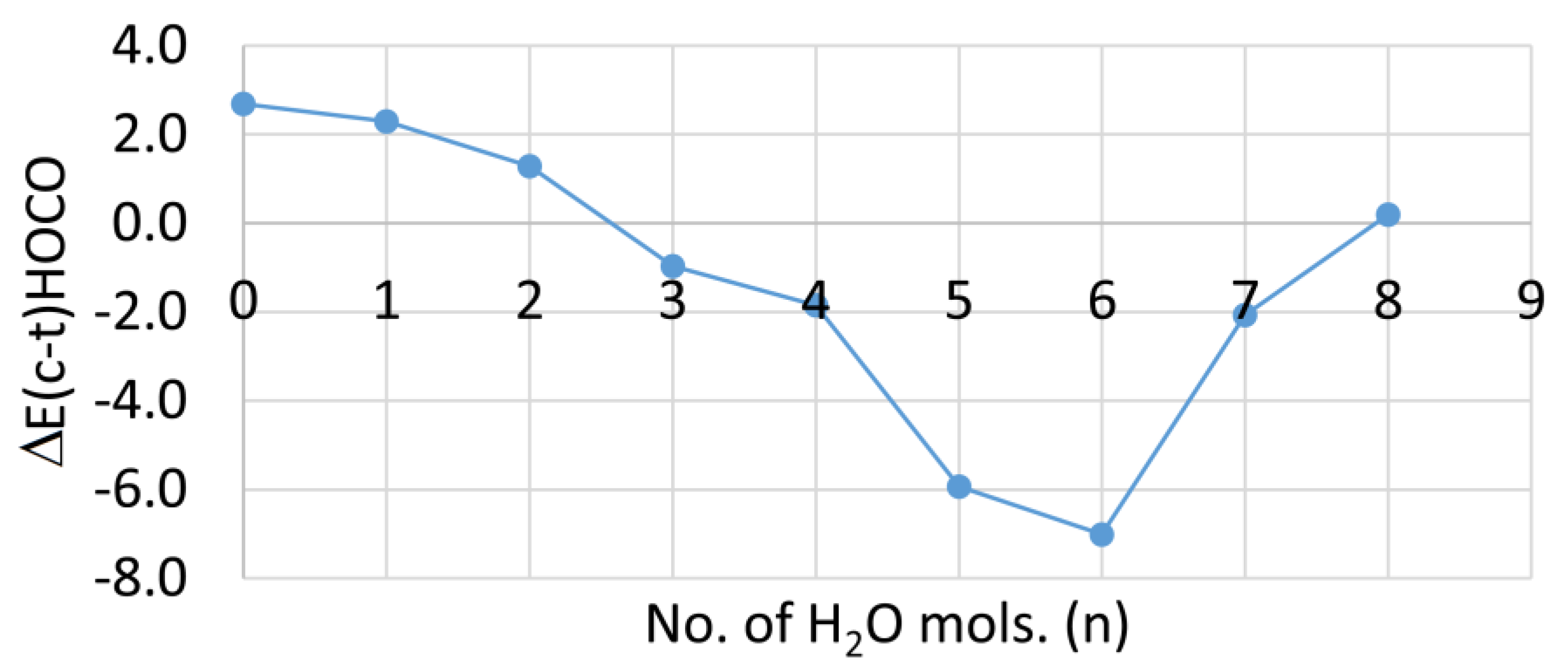
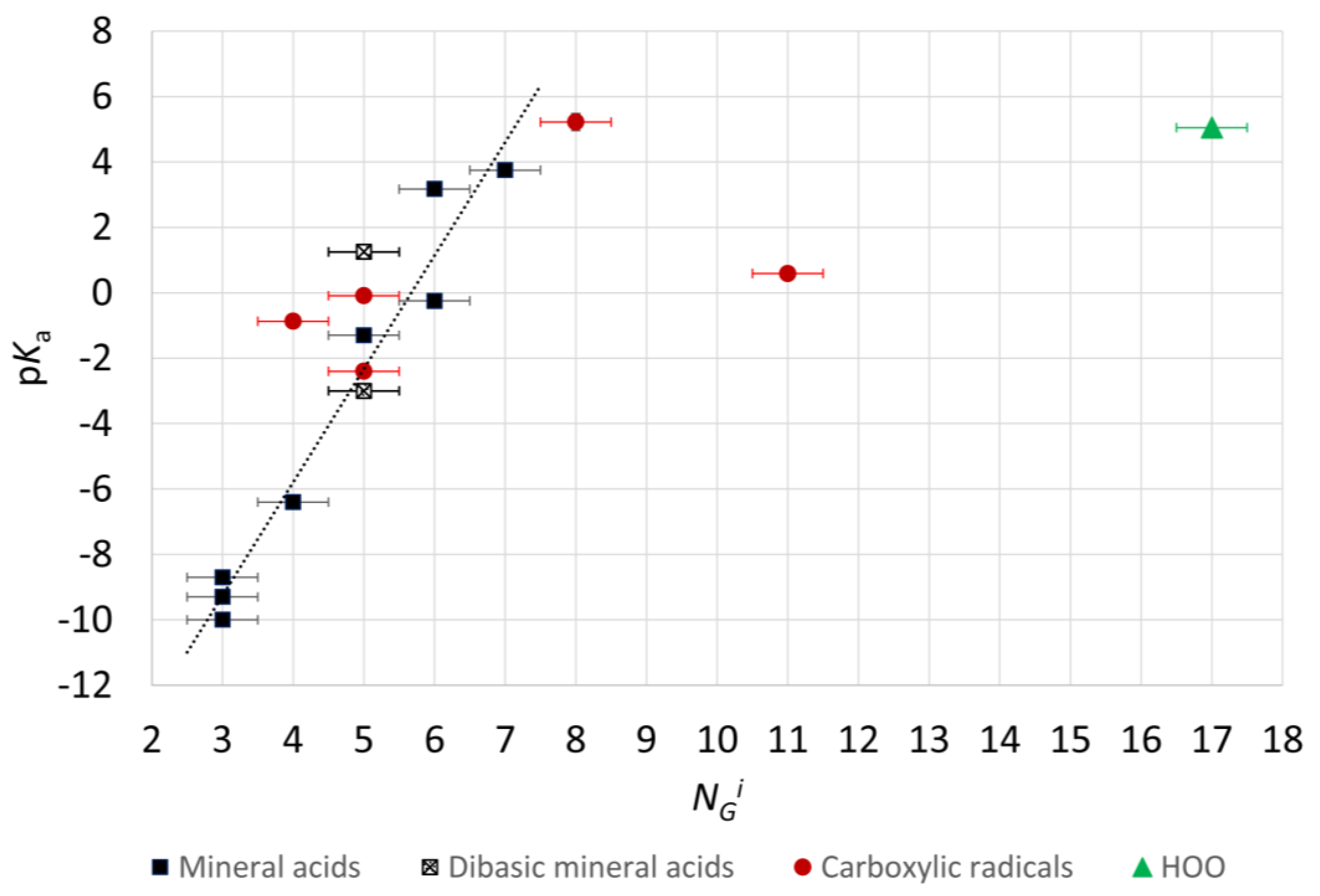
2.4. Microhydration, Acidity, and RED-Shift
3. Materials and Methods
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Hayon, E.; Simic, M. Acid-base Properties of Free Radicals in Solution. Acc. Chem. Res. 1974, 7, 114–121. [Google Scholar] [CrossRef]
- Steenken, S.; Telo, J.P.; Novais, H.M.; Candeias, L.P. One-electron-reduction Potentials of Pyrimidine Bases, Nucleosides, and Nucleotides in Aqueous Solution. Consequences for DNA redox Chemistry. J. Am. Chem. Soc. 1992, 114, 4701–4709. [Google Scholar] [CrossRef]
- Steenken, S.; Neta, P. One-electron Redox Potentials of Phenols. Hydroxy- and Aminophenols and Related Compounds of Biological Interest. J. Phys. Chem. 1982, 86, 3661–3667. [Google Scholar] [CrossRef]
- Buckel, W.; Keese, R. One-electron Redox Reactions of CoASH Esters in Anaerobic Bacteria—A Mechanistic Proposal. Angew. Chem. Int. Ed. 1995, 34, 1502–1506. [Google Scholar] [CrossRef]
- Smith, D.M.; Buckel, W.; Zipse, H. Deprotonation of Enoxy Radicals: Theoretical Validation of a 50-Year-Old Mechanistic Proposal. Angew. Chem. Int. Ed. 2003, 42, 1867–1870. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Darley, D.J.; Buckel, W.; Pierik, A.J. An Allylic Ketyl Radical Intermediate in Clostridial Amino-acid Fermentation. Nat. Chem. 2008, 44, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Curran, D.P. The Electron is a Catalyst. Nat. Chem. 2014, 50, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Mayer, P.M.; Radom, L. Deprotonating Molecules and Free Radicals to Form Carbon-Centered Anions: A G2 ab Initio Study of Molecular and Free Radical Acidity. J. Phys. Chem. A 1998, 102, 4918–4924. [Google Scholar] [CrossRef]
- Morris, M.; Chan, B.; Radom, L. Effect of Protonation State and Interposed Connector Groups on Bond Dissociation Enthalpies of Alcohols and Related Systems. J. Phys. Chem. A 2014, 118, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.C. Radical-Enhanced Acidity: Why Bicarbonate, Carboxyl, Hydroperoxyl, and Related Radicals Are So Acidic. J. Phys. Chem. A 2017, 121, 7761–7767. [Google Scholar] [CrossRef] [PubMed]
- Schmidt am Busch, M.; Knapp, E.-W. Accurate pKa Determination for a Heterogeneous Group of Organic Molecules. ChemPhysChem 2004, 5, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.O.; da Silva, E.C.; Nascimento, M.A.C. Ab initio Calculations of Absolute pKa Values in Aqueous Solution. Part 2. Aliphatic Alcohols, Thiols, and Halogenated Carboxylic Acids. J. Phys. Chem. A 2000, 104, 2402–2409. [Google Scholar] [CrossRef]
- Klicic, J.J.; Freisner, R.A.; Liu, S.-Y.; Guida, W.C. Accurate Prediction of Acidity Constants in Aqueous Solution via Density Functional Theory and Self-Consistent Reaction Field Methods. J. Phys. Chem. A 2002, 106, 1327–1335. [Google Scholar] [CrossRef]
- Klamt, A.; Eckert, F.; Diedenhofen, M.; Beck, M.E. First Principles Calculations of Aqueous pKa Values for Organic and Inorganic Acids Using COSMO-RS Reveal an Inconsistency in the Slope of the pKa Scale. J. Phys. Chem. A 2003, 107, 9380–9386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Baker, J.; Pulay, P.A. Reliable and Efficient First Principles-Based Method for Predicting pKa Values. 1. Methodology. J. Phys. Chem. A 2010, 114, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Baker, J.; Pulay, P.A. Reliable and Efficient First Principles-Based Method for Predicting pKa Values. 2. Organic Acids. J. Phys. Chem. A 2010, 114, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; DaBell, P.; Manley, D.W.; McCaughan, R.P.; Walton, J.C. Bicarbonate and Alkyl Carbonate Radicals: Structural Integrity and Reactions with Lipid Components. J. Am. Chem. Soc. 2015, 137, 16153–16162. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.C. Enhanced Proton Loss from Neutral Free Radicals: Towards Carbon-Centered Superacids. J. Phys. Chem. A 2018, 122. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Gutberlet, A.; Schwaab, G.; Birer, O.; Masia, M.; Kaczmarek, A.; Forbert, H.; Havenith, M.; Marx, D. Science 2009, 324, 1545–2212. [CrossRef] [PubMed]
- Leopold, K.R. Hydrated Acid Clusters. Annu. Rev. Phys. Chem. 2011, 62, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.H.; Tao, F.M. Ionic Dissociation of Perchloric Acid in Microsolvated Clusters. J. Phys. Chem. A 2001, 105, 1208–1213. [Google Scholar] [CrossRef]
- Del Valle, C.P.; Valdemoro, C.; Novoa, J.J. The Determinant Role of Water in the Ionic Dissociation of HO2. J. Mol. Struct. THEOCHEM 1996, 371, 143–152. [Google Scholar] [CrossRef]
- Close, D.M.; Wardman, P. Calculation of Standard Reduction Potentials of Amino Acid Radicals and the Effects of Water and Incorporation into Peptides. J. Phys. Chem. A 2018, 122, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 Theory. J. Chem. Phys. 2007, 126, 84108–84119. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-correlation Functional Using the Coulomb-attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Maity, D.K. How Much Water Is Needed To Ionize Formic Acid? J. Phys. Chem. A 2013, 117, 8660–8670. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, P.; Maity, D.K. Effect of microhydration on dissociation of trifluoroacetic acid. J. Phys. Chem. A 2014, 118, 5443–5453. [Google Scholar] [CrossRef] [PubMed]
- Jeevarajan, A.S.; Carmichael, I.; Fessenden, R.W. ESR Measurement of the pKa of Carboxyl Radical and ab initio Calculation of the Carbon-13 Hyperfine Constant. J. Phys. Chem. 1990, 94, 1372–1376. [Google Scholar] [CrossRef]
- Forney, D.; Jacox, M.E.; Thompson, W.E. Infrared Spectra of trans-HOCO, HCOOH+, and HCO2− Trapped in Solid Neon. J. Chem. Phys. 2003, 119, 10814–10823. [Google Scholar] [CrossRef]
- Johnson, C.J.; Otto, R.; Continetti, R.E. Spectroscopy and Dynamics of the HOCO Radical: Insights into the OH + CO → H + CO2 Reaction. Phys. Chem. Chem. Phys. 2014, 16, 19091–19105. [Google Scholar] [CrossRef] [PubMed]
- Ryazantsev, S.V.; Feldman, V.I.; Khriachtchev, L. Conformational Switching of HOCO Radical: Selective Vibrational Excitation and Hydrogen-Atom Tunneling. J. Am. Chem. Soc. 2017, 139, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Radford, H.E.; Wei, W. The rotational spectrum of trans-HOCO and DOCO. J. Chem. Phys. 1992, 97, 3989–3995. [Google Scholar] [CrossRef]
- Oyama, T.; Funato, W.; Sumiyoshi, Y.; Endoa, Y. Observation of the Pure Rotational Spectra of trans- and cis-HOCO. J. Chem. Phys. 2011, 134, 174303–174307. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.C.; Martinez, O.; McGuire, B.A.; Crabtree, K.N.; Martin-Drumel, M.-A.; Stanton, J.F. Isotopic Studies of trans- and cis-HOCO Using Rotational Spectroscopy: Formation, Chemical Bonding, and Molecular Structures. J. Chem. Phys. 2016, 144, 124304. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Francisco, J.S. High Level ab initio Studies on the Excited States of HOCO Radical. J. Chem. Phys. 2000, 113, 7963–7970. [Google Scholar] [CrossRef]
- Conte, R.; Houston, P.L.; Bowman, J.M. Communication: A Benchmark-quality, Full-dimensional ab initio Potential Energy Surface for Ar-HOCO. J. Chem. Phys. 2014, 140, 151101. [Google Scholar] [CrossRef]
- As a cross check suggested by a reviewer, structures of the 4×H2O and 5×H2O clusters of 2H were optimized with the CAM-B3LYP-D3/6-311+G(2d,p) method to include Grimme’s empirical dispersion corrections. The SMD solvent continuum model was also employed. For each a range of different starting geometries was tried. For both cluster types this method arrived at the same global minimum structures 2H.4H2O and 2H.5H2O shown in Figure 5. There were some differences in bond lengths and angles. Furthermore, the cluster of the 2H.5H2O system exhibiting H-bonding to the terminal atom of the ethyne unit as part of a ring connected to a carboxyl O-atom (Figure 5) was again found to be only a local minimum but 7 kcal/mol higher in energy.
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O2− Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Mendes, J.; Zhou, C.-W.; Curran, H.J. Theoretical Chemical Kinetic Study of the H-Atom Abstraction Reactions from Aldehydes and Acids by H Atoms and OH, HO2, and CH3 Radicals. J. Phys. Chem. A 2014, 118, 12089–12104. [Google Scholar] [CrossRef] [PubMed]
- Farnia, S.; Vahedpour, M.; Abedi, M.; Farrokhpour, H. Theoretical Study on the Mechanism and Kinetics of Acetaldehyde and Hydroperoxyl Radical: An Important Atmospheric Reaction. Chem. Phys. Lett. 2013, 583, 190–197. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Russo, N. Antioxidant Activity of trans-Resveratrol toward Hydroxyl and Hydroperoxyl Radicals: A Quantum Chemical and Computational Kinetics Study. J. Org. Chem. 2012, 77, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Garcia, J. Theoretical Study of the Trapping of the OOH Radical by Coenzyme Q. J. Am. Chem. Soc. 2004, 126, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Torrent-Sucarrat, M.; Ruiz-Lopez, M.F.; Martins-Costa, M.; Francisco, J.S.; Anglada, J.M. Protonation of Water Clusters Induced by Hydroperoxyl Radical Surface Adsorption. Chem. Eur. J. 2011, 17, 5076–5085. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Tawada, Y.; Tsuneda, T.; Yunagisawa, S.; Yanai, T.; Hirao, K. A Long-Range-Corrected Time-Dependent Density Functional Theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
Structure Availability: Cartesian matrices of the optimum clusters are available from the author. |










© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, J.C. Microhydration and the Enhanced Acidity of Free Radicals. Molecules 2018, 23, 423. https://doi.org/10.3390/molecules23020423
Walton JC. Microhydration and the Enhanced Acidity of Free Radicals. Molecules. 2018; 23(2):423. https://doi.org/10.3390/molecules23020423
Chicago/Turabian StyleWalton, John C. 2018. "Microhydration and the Enhanced Acidity of Free Radicals" Molecules 23, no. 2: 423. https://doi.org/10.3390/molecules23020423
APA StyleWalton, J. C. (2018). Microhydration and the Enhanced Acidity of Free Radicals. Molecules, 23(2), 423. https://doi.org/10.3390/molecules23020423