Recognition of AMP, ADP and ATP through Cooperative Binding by Cu(II) and Zn(II) Complexes Containing Urea and/or Phenylboronic—Acid Moieties

 , ,
, ,
Abstract
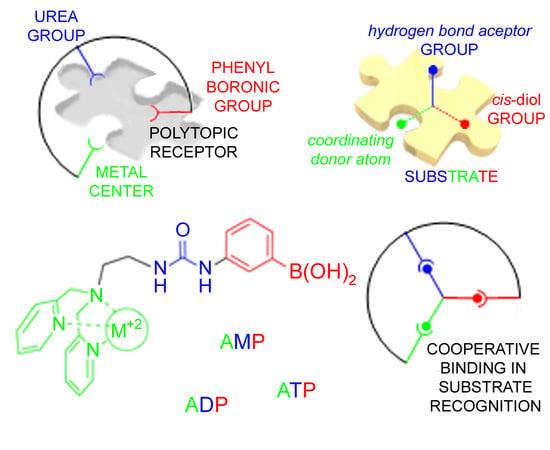
:
1. Introduction
2. Results
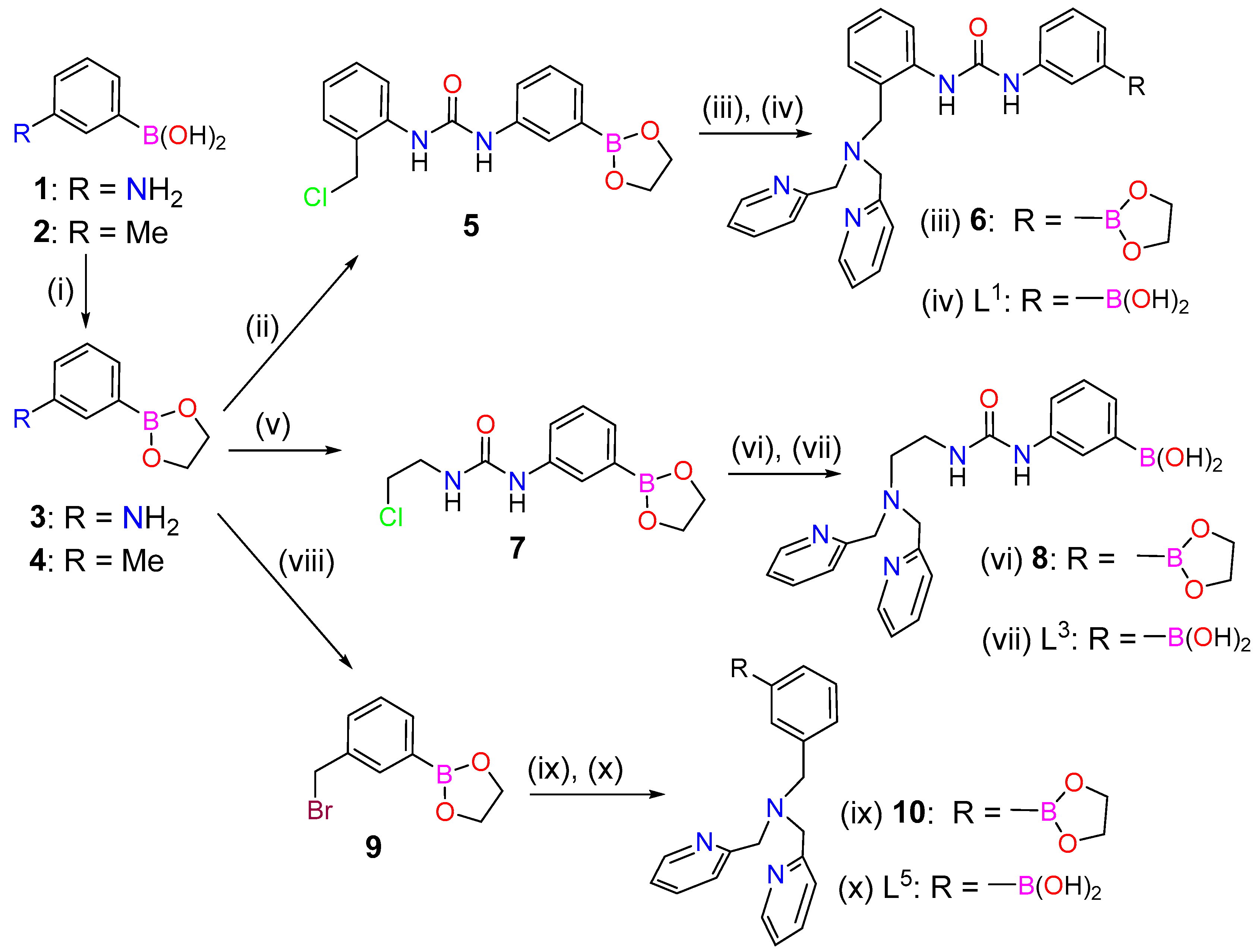
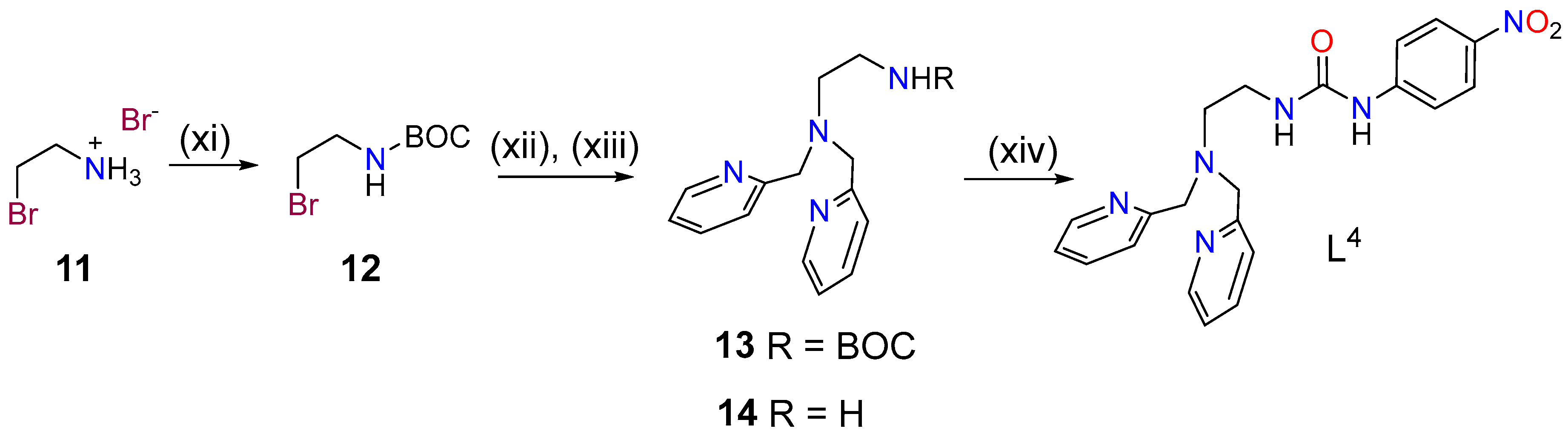
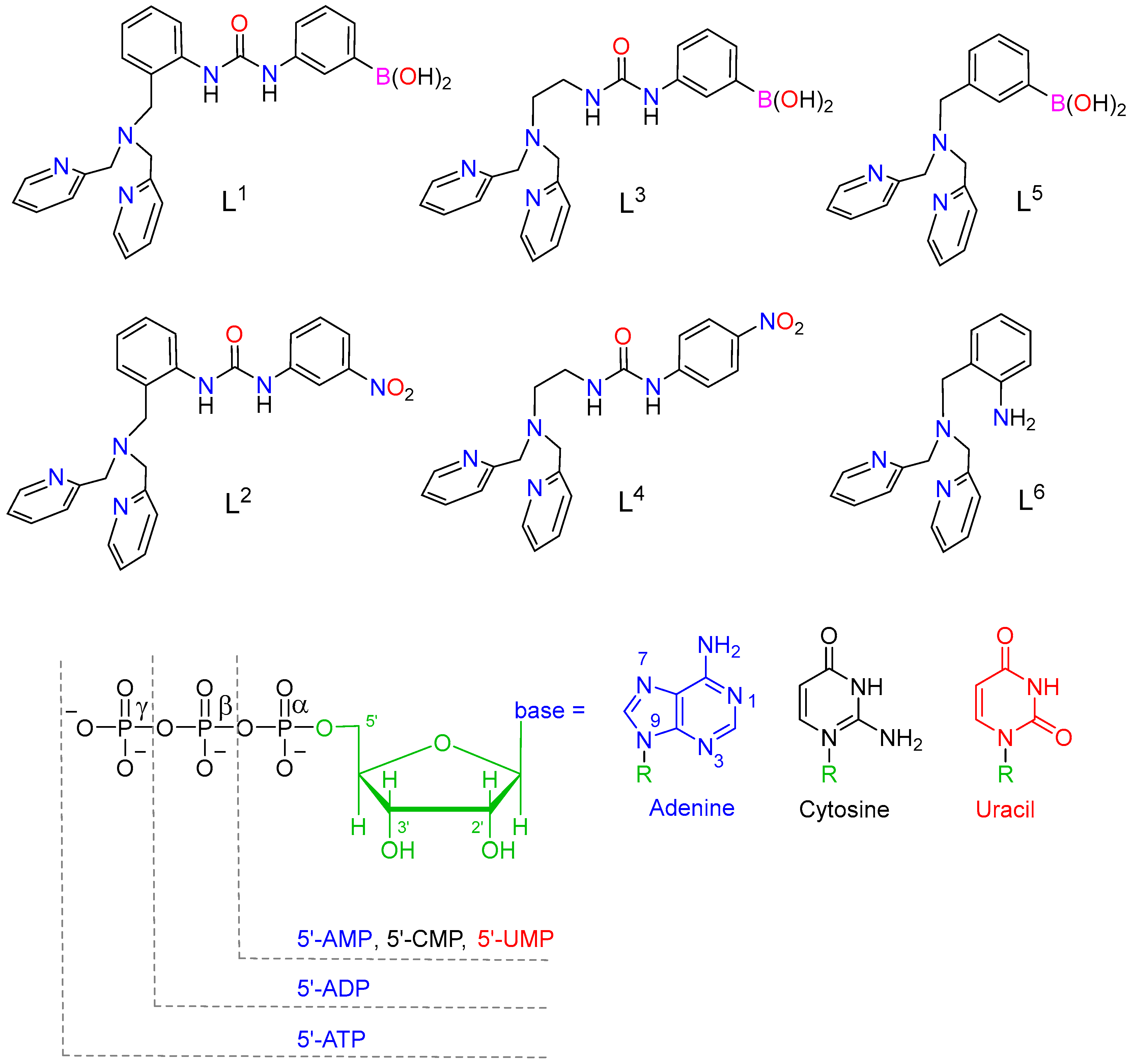
2.1. Synthesis and Characterization of the Ligands and Metal Complexes
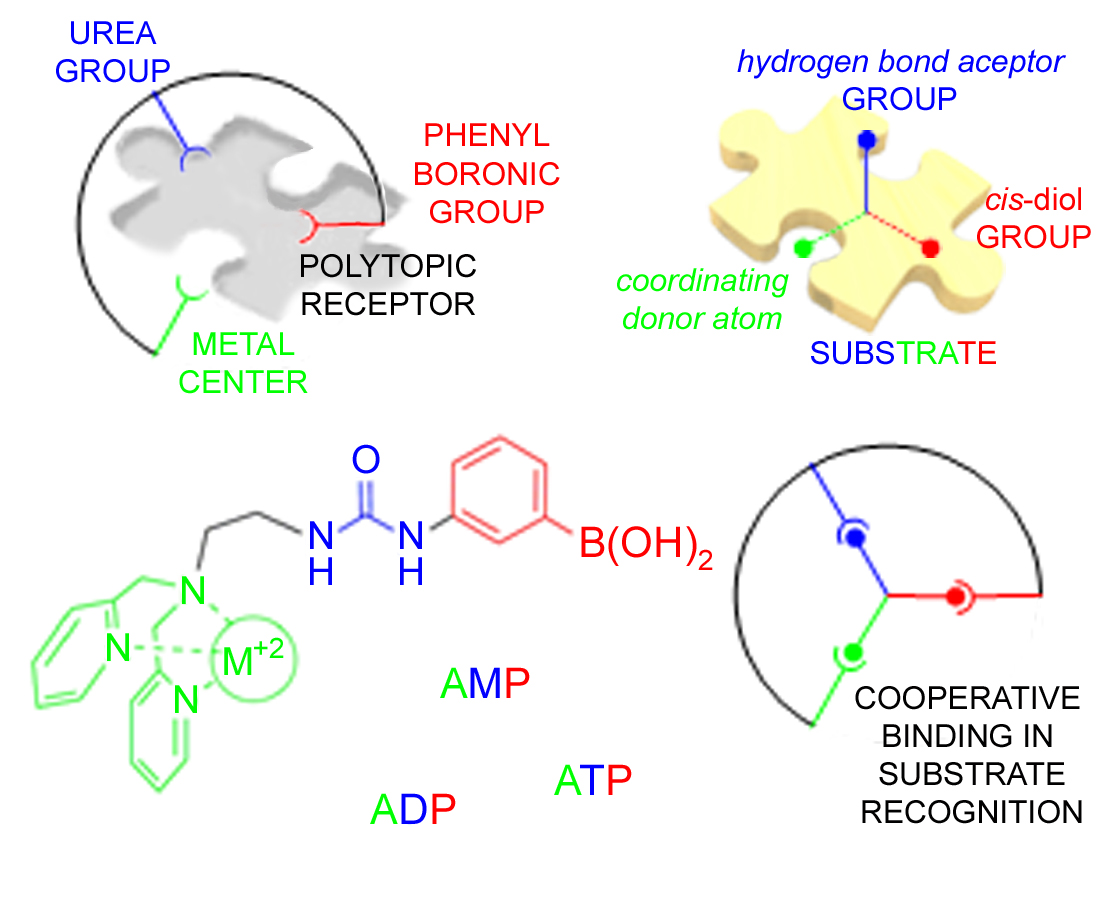
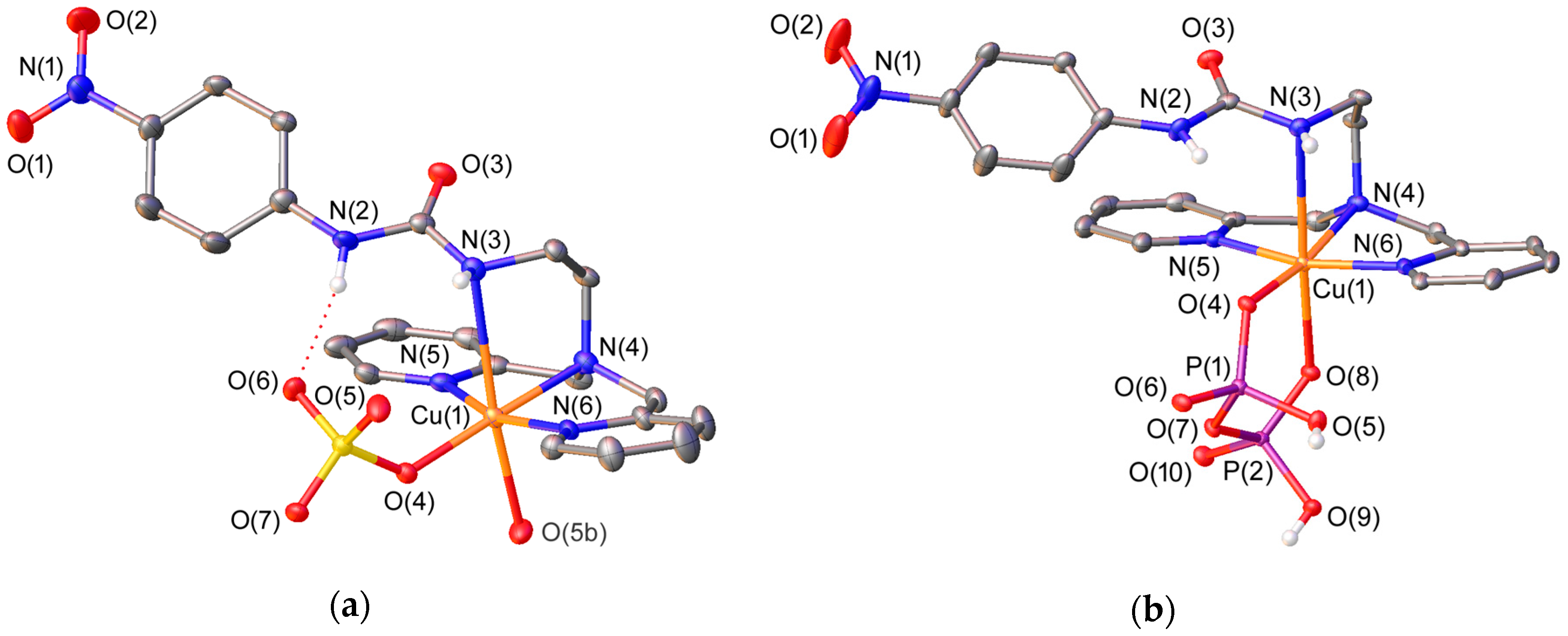
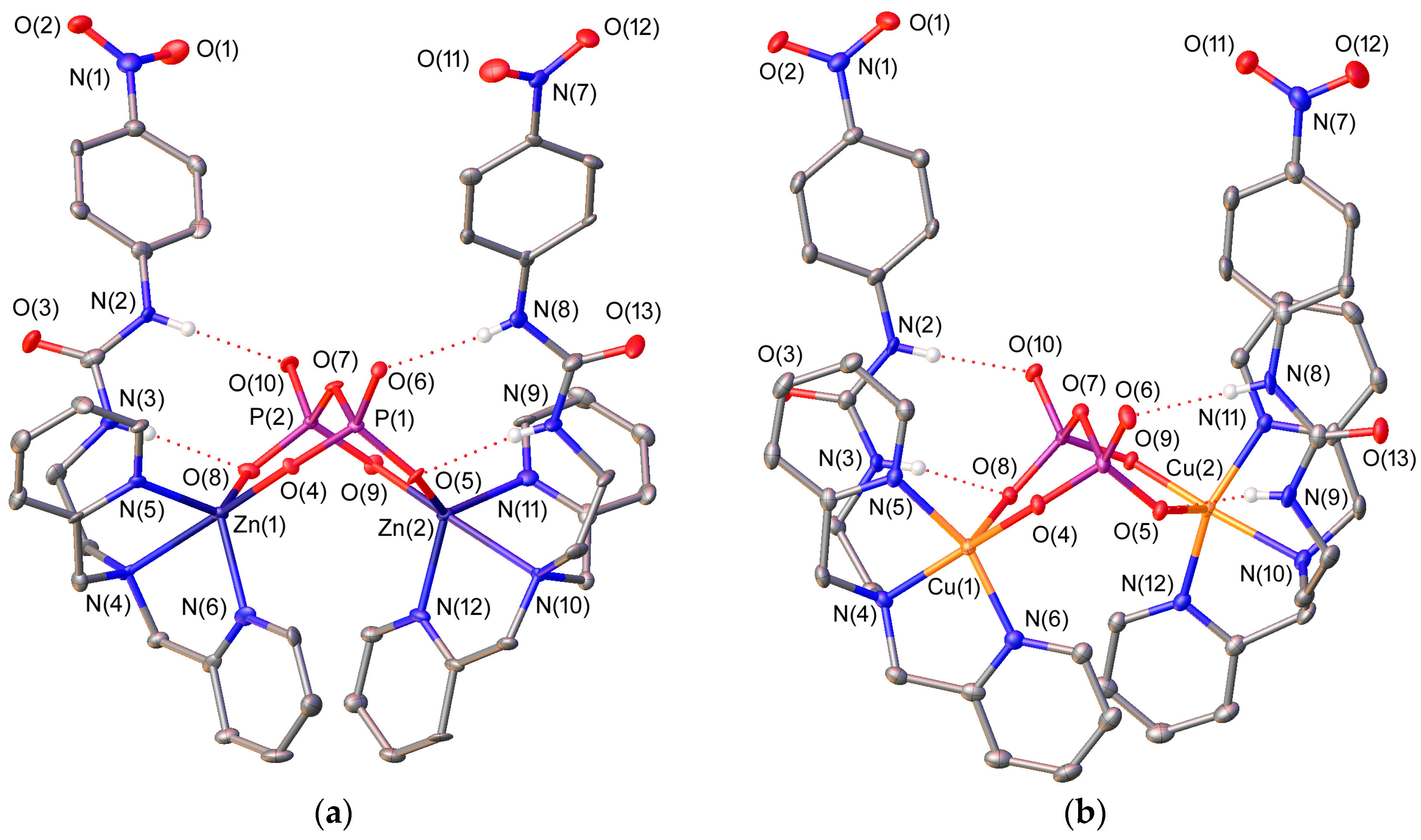
2.2. X-ray Crystal Structures
2.3. Stability of the Complexes Toward Hydrolysis
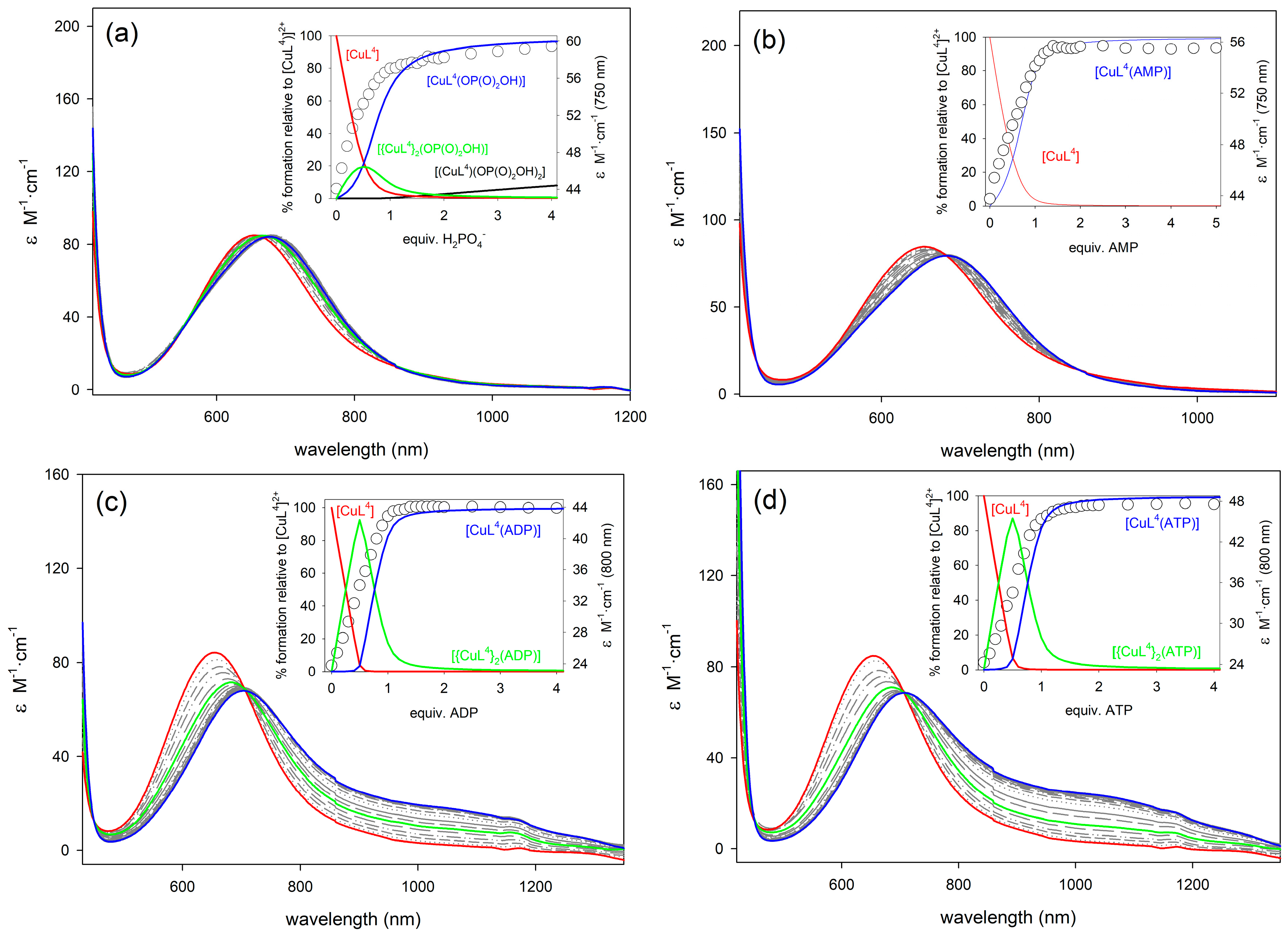
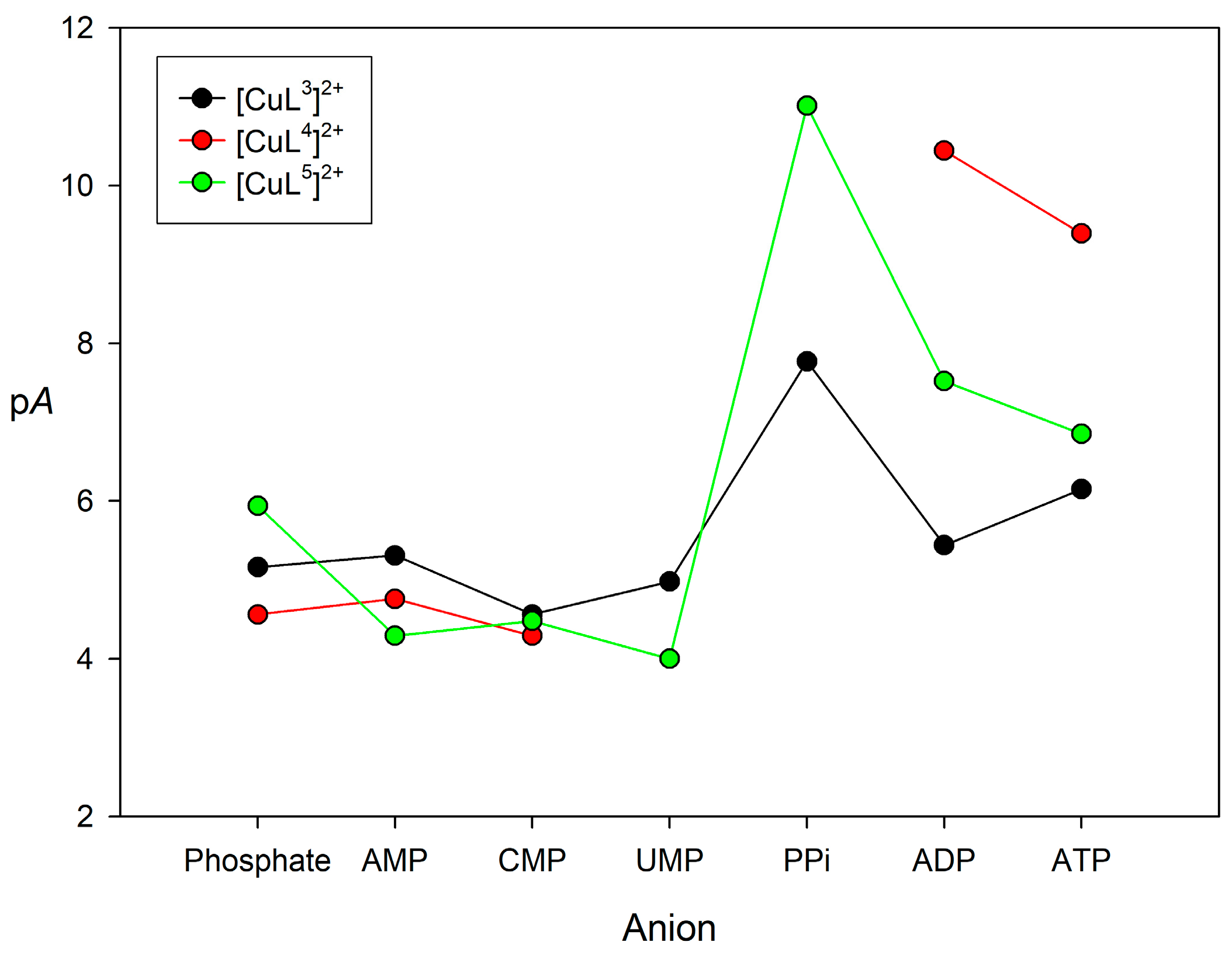
2.4. Binding to Phosphorylated Anions
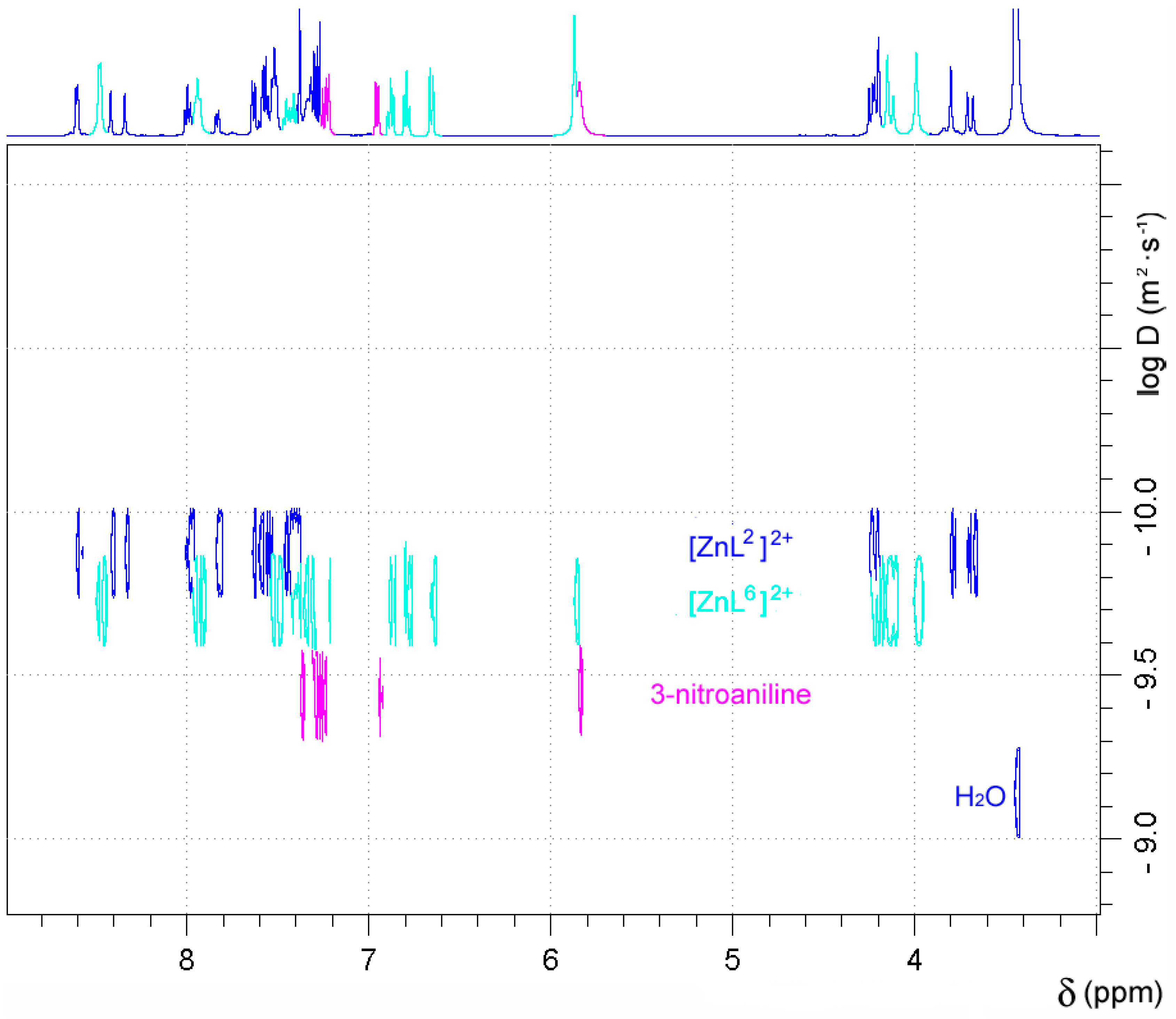
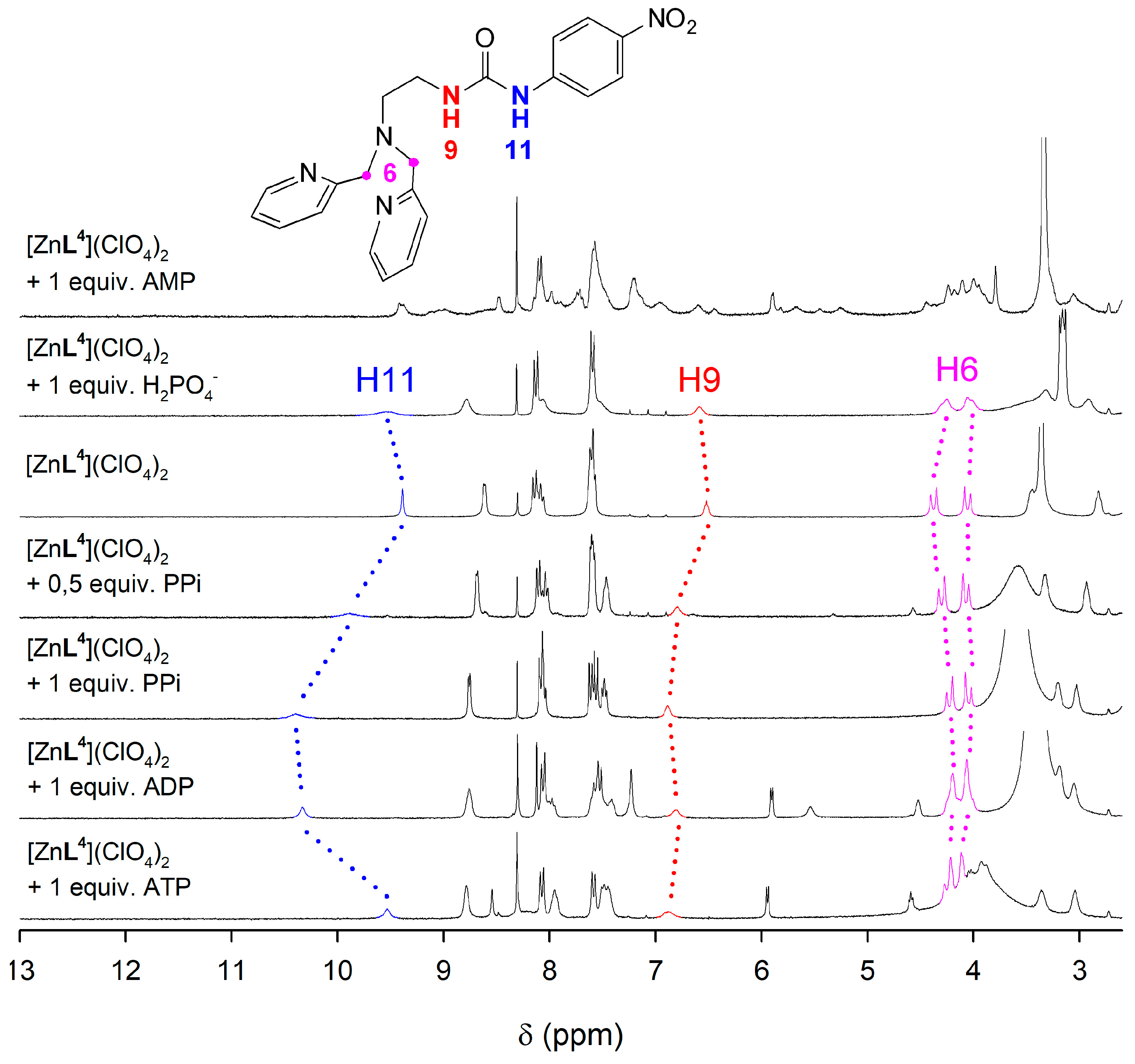
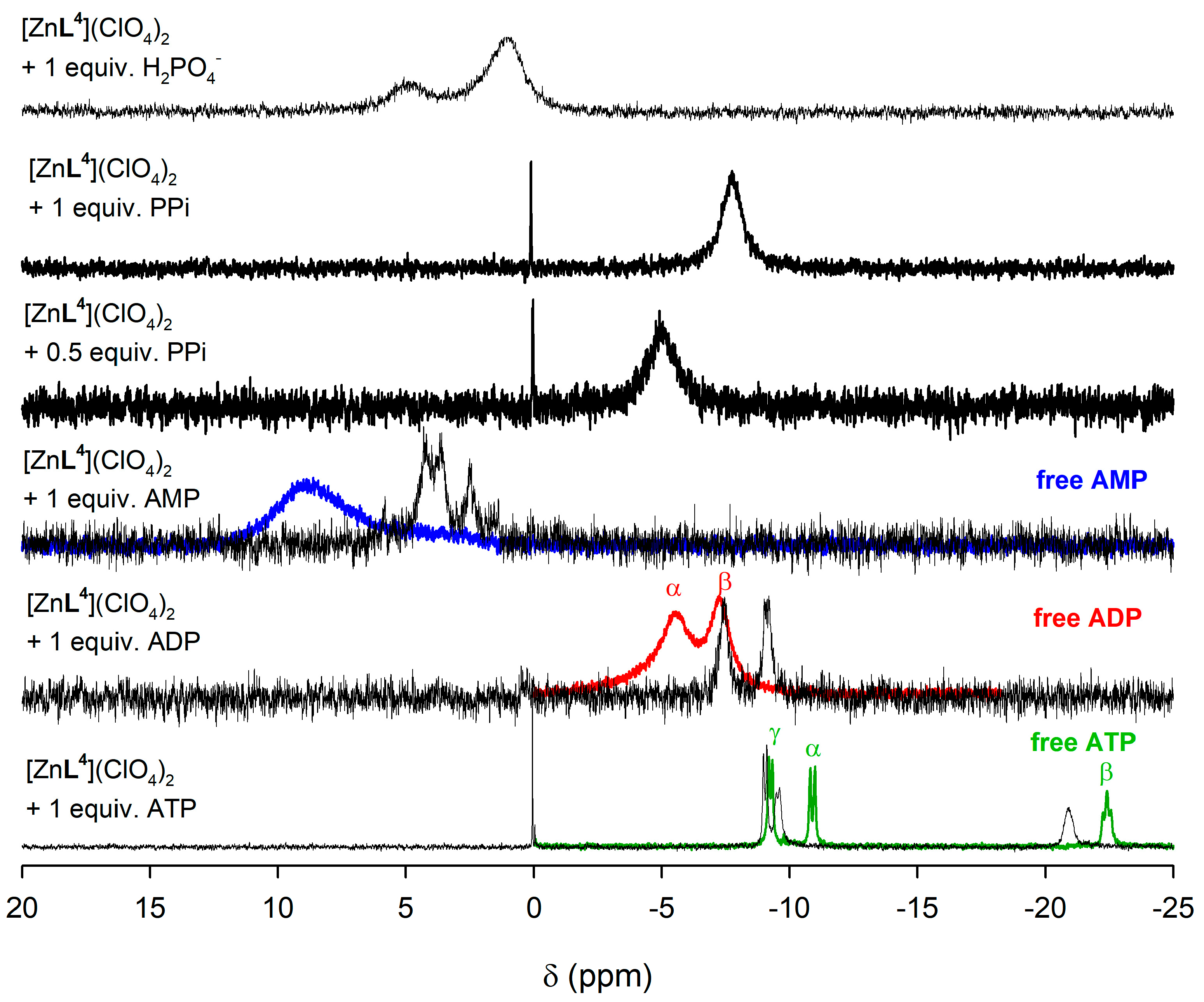
2.5. NMR Studies
3. Materials and Methods
3.1. General Methods
Reagents
3.2. Synthesis
3.3. X-ray Diffraction Studies
3.4. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hargrove, A.E.; Nieto, S.; Zhang, T.; Sessler, J.L.; Anslyn, E.V. Artificial Receptors for the Recognition of Phosphorylated Molecules. Chem. Rev. 2011, 111, 6603–6782. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.R. Enzyme-Catalyzed Phosphoryl Transfer Reactions. Annu. Rev. Biochem. 1980, 49, 877–919. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic cotransmission. Exp. Physiol. 2009, 94, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A. AMP-activated protein kinase: The energy charge hypothesis revisited. BioEssay 2001, 23, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Suprenant, A.; Evans, R.J. ATP in synapses. Nature 1993, 362, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Murugappa, S.; Kunapuli, S.P. The role of ADP receptors in platelet function. Front. Biosci. 2006, 11, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Rehamn, G.; Shehzad, A.; Khan, A.L.; Hamayun, M. Role of AMP-Activated Protein Kinase in Cancer Therapy. Arch. Pharm. Chem. Life Sci. 2014, 347, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Liang, L.; Zhang, L. Ratiometric Fluorescence Recognition for Pyrophosphate on the Basis of Terpyridine Derivative. Anal. Sci. 2013, 29, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Wang, H.; Gao, X.D.; Yang, R.H.; Chan, W.H. Spiropyran-based fluorescent anion probe and its application for urinary pyrophosphate detection. Anal. Chem. 2010, 82, 4628–4636. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.-S.; Bae, S.W.; Kim, H.; Hong, J.-I. Electrogenerated chemiluminescent anion sensing: Selective recognition and sensing of pyrophosphate. Anal. Chem. 2010, 82, 8259–8265. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yuen, K.K.Y.; Jolliffe, K.A.; Yoon, J. Fluorescent and Colorimetric Chemosensors for Pyrophosphate. Chem. Soc. Rev. 2015, 44, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Wu, S.-M.; Tseng, W.-L. Magnetite Nanoparticle-Induced Fluorescence Quenching of Adenosine Triphosphate−BODIPY Conjugates: Application to Adenosine Triphosphate and Pyrophosphate Sensing. Anal. Chem. 2013, 85, 8559–8565. [Google Scholar] [CrossRef] [PubMed]
- McDonough, M.J.; Reynolds, A.J.; Lee, W.Y.G.; Jolliffe, K.A. Selective recognition of pyrophosphate in water using a backbone modified cyclic peptide receptor. Chem. Commun. 2006, 2971–2973. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Reddy, M.D.; Mishra, A.; George, S.J. The molecular recognition controlled stereomutation cycle in a dynamic helical assembly. Org. Biomol. Chem. 2015, 13, 9938–9942. [Google Scholar] [CrossRef] [PubMed]
- Francesconi, O.; Gentili, M.; Bartoli, F.; Bencini, A.; Conti, L.; Giorgi, C.; Roelens, S. Phosphate binding by a novel Zn(II) complex featuring a trans-1,2-diaminocyclohexane ligand. Effective anion recognition in water. Org. Biomol. Chem. 2015, 13, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yu, P.; Yang, L.; Mao, L. Competitive Coordination of Cu2+ between Cysteine and Pyrophosphate Ion: Toward Sensitive and Selective Sensing of Pyrophosphate Ion in Synovial Fluid of Arthritis Patients. Anal. Chem. 2015, 85, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Ying, Y.-L.; Hua, X.; Niu, Y.; Xu, Y.; Long, Y.-T. Electrochemically generated green-Fluorescent N-doped carbon quantum dots for facile monitoring alkaline phosphatase activity based on the Fe3+-mediating ON-OFF-ON-OFF fluorescence principle. Carbon 2018, 127, 340–348. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, J.; Wu, X.; Lin, L.; Weng, S.; Chen, M.; Cai, X.; Lin, X. Switch-on fluorescent strategy based on N and S co-doped graphene quantum dots (N-S/GQDs) for monitoring pyrophosphate ions in synovial fluid of arthritis patients. Sens. Actuators B 2016, 229, 217–224. [Google Scholar] [CrossRef]
- Smith, B.A.; Akers, W.J.; Leevy, W.M.; Lampkins, A.J.; Xiao, S.; Wolter, W.; Suckow, M.A.; Achilefu, S.; Smith, B.D. Optical Imaging of Mammary and Prostate Tumors in Living Animals using Synthetic Near Infrarred Zinc(II)-Dipicolylamine Probe for Anionic Cell Surfaces. J. Am. Chem. Soc. 2010, 132, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.R.; Lupták, A. DNA synthesis from diphosphate substrates by DNA polymerases. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Emami, F.; Nishiyabu, R.; Kubo, Y.; Anzenbacher, P., Jr. Quantitative analysis of modeled ATP hydrolysis in water by a colorimetric sensor array. Chem. Commun. 2016, 52, 7838–7841. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, K.A. Pyrophosphate Recognition and Sensing in Water Using Bis[zinc(II)dipicolylamino]-Functionalized Peptides. Acc. Chem. Res. 2017, 50, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Sigel, H.; Massoud, S.S.; Corfù, N.A. Comparison of the Extent of Macrochelate Formation in Complexes of Divalent Metal Ions with Guanosine (GMP2−), Inosine (IMP2−), and Adenosine 5′-Monophosphate (AMP2−). The Crucial Role of N-7 Basicity in Metal Ion-Nucleic Base Recognition. J. Am. Chem. Soc. 1994, 116, 2958–2971. [Google Scholar] [CrossRef]
- Sigel, H.; Massoud, S.S.; Tribolet, R. Comparison of the Metal Ion Coordinating Properties of Tubercidin 5′-Monophosphate (7-Deaza-AMP) with Those of Adenosine 5′-Monophosphate (AMP) ans 1,N6-Ethenoadenosine 5′-Monophosphate (∈-AMP). Definite Evidence for Metal Ion-Base Backbinding to N-7 and Extent of Macrochelate Formation in M(AMP) and M(∈-AMP). J. Am. Chem. Soc. 1988, 110, 6857–6865. [Google Scholar] [CrossRef]
- Massoud, S.S.; Tribolet, R.; Sigel, H. Metal-ion-governed molecular recognition: extent of intramolecular stack formation in mixed-ligand-copper(II) complexes containing a heteroaromatic N base and an adenosine monophosphate (2′AMP, 3′AMP, or 5′AMP) A structuring effect of the metal-ion bridge. Eur. J. Biochem. 1990, 187, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Sigel, H. Interactions of Metal Ions with Nucleotides and Nucleic Acids and their Constituents. Chem. Soc. Rev. 1993, 22, 255–267. [Google Scholar] [CrossRef]
- Massoud, S.S.; Sigel, H. Evaluation of the metal-ion-coordinating differences between the 2′-, 3′- and 5′-monophosphates of adenosine. Eur. J. Biochem. 1989, 179, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Massoud, S.S.; Sigel, H. Metal Ion Coordinating Properties of Pyrimidine-Nucleoside 5′-Monophosphates (CMP, UMP, TMP) and of Simple Phosphate Monoesters, Including D-Ribose 5′-Monophosphate. Establishment of Relations between Complex Stability and Phosphate Basicity. Inorg. Chem. 1988, 27, 1447–1453. [Google Scholar] [CrossRef]
- Aoki, K. X-ray evidence for the metal ion bridged intra- and intermolecular stacking interactions between nucleotide bases and aromatic heterocyclic rings within the ternary complex [Cu(5′-AMP)(bpy)(H2O)]2(NO3)2·6H2O. J. Am. Chem. Soc. 1978, 100, 7106–7108. [Google Scholar] [CrossRef]
- Pouessel, J.; Abada, S.; Le Bris, N.; Elhabiri, M.; Charbonnière, L.J.; Tripier, R. A new bis-tetraamine ligand with a chromophoric 4-(9-anthracenyl)-2,6-dimethylpyridinyl linker for glyphosate and ATP sensing. Dalton Trans. 2013, 42, 4859–4872. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, J.; Bazzicalipi, C.; Bencini, A.; Bernard, H.; Giorgi, C.; Handel, H.; Matera, I.; Le Bris, N.; Tripier, R.; Valtancoli, B. Exploring New Molecular Architectures for Anion Recognition: Synthesis and ATP Binding Properties of New Cyclam-Based Ditopic Polyammonium Receptors. Chem. Asian J. 2011, 6, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S. Receptors for Biologically Relevant Anions. In Anion Coordination Chemistry; Bowman-James, K., Bianchi, A., García-España, E., Eds.; John Wiley & Sons: Weinheim, Germany, 2012; pp. 363–464. ISBN 978-3-527-32370-8. [Google Scholar]
- Bregier-Jarzebowska, R.; Gasowska, A.; Hoffman, S.K.; Lomozik, L. Interactions of diamines with adenosine-5′-triphosphate (ATP) in the systems including copper(II) ions. J. Inorg. Biochem. 2006, 162, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kataev, E.A.; Müller, C.; Kolesnikov, G.V.; Khrustalev, V.N. Guanidinium-Based Artificial Receptors for Binding Orthophosphate in Aqueous Solution. Eur. J. Org. Chem. 2014, 2747–2753. [Google Scholar] [CrossRef]
- Xu, Z.; Singh, N.J.; Lim, J.; Pan, J.; Kim, H.-N.; Park, S.; Kim, K.-S.; Yoon, J. Unique Sandwich Stacking of Pyrene-Adenine-Pyrene for Selective and Ratiometric Fluorescent Sensing of ATP at Physiological pH. J. Am. Chem. Soc. 2009, 131, 15528–15533. [Google Scholar] [CrossRef] [PubMed]
- Xuejian, L.; Smith, D.G.; Jolliffe, K.A. Are two better than one? Comparing intermolecular and intramolecular indicator displacement assays in pyrophosphate sensors. Chem. Commun. 2016, 52, 8463–8466. [Google Scholar] [CrossRef]
- Zwicker, V.E.; Liu, X.; Yuen, K.K.Y.; Jolliffe, K.A. Triazole-containing zinc(II)dipicolylamine-functionalised peptides as highly selective pyrophosphate sensors in physiological media. Supramol. Chem. 2016, 28, 192–200. [Google Scholar] [CrossRef]
- Zhao, R.R.; Xu, Q.L.; Yang, Y.; Cao, J.; Zhou, Y.; Xu, R.; Zhang, J.F. A coumarin-based terpyridine-zinc complex for sensing pyrophosphate and its application in in vivo imaging. Tetrahedron Lett. 2016, 57, 5022–5025. [Google Scholar] [CrossRef]
- Chao, D.; Ni, S. Nanomolar pyrophosphate detection and nucleus staining in living cells with simple terpyridine-Zn(II) complexes. Sci. Rep. 2016, 6, 26477. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chandar, N.B.; Chourey, S.; Agarwalla, H.; Ganguly, B.; Das, A. Role of Metal Ion in Specific Recognition of Pyrophosphate Ion under Physiological Conditions and Hydrolysis of the Phosphoester Linkage by Alkaline Phosphatase. Inorg. Chem. 2013, 52, 11034–11041. [Google Scholar] [CrossRef] [PubMed]
- Watchasit, S.; Suktanarak, P.; Suksai, C.; Ruangpornvisuti, V.; Tuntulani, T. Discriminate sensing of pyrophosphate using a new tripodal tetramine-based dinuclear Zn(II) complex under an indicator displacement assay approach. Dalton Trans. 2014, 43, 14701–14709. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hao, G.; Wu, F.; Feng, G. Fluorescence sensing of ADP over ATP and PPi in 100% aqueous solution. Analyst 2015, 140, 5873–5876. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pandey, R.; Kumar, A.; Gupta, R.K.; Dubey, M.; Mohammed, A.; Mobin, S.M.; Pandey, D.S. Self-assembled copper(II) metallacycles derived from asummetric Schiff base ligands: Efficient hosts for ADP/ATP in phosphate buffer. Dalton Trans. 2015, 44, 17152–17165. [Google Scholar] [CrossRef] [PubMed]
- Mesaquita, L.M.; Mateus, P.; Fernandez, R.D.V.; Iranzo, O.; André, V.; de Oliveira, F.T.; Platas-Iglesias, C.; Delgado, R. Recognition of phosphopeptides by a dinuclear copper(II) macrocyclic complex in a water: Methanol 50:50 v/v solution. Dalton Trans. 2017, 46, 9549–9564. [Google Scholar] [CrossRef] [PubMed]
- Amendola, V.; Bergamaschi, G.; Guglielmo, L.; Izzo, L.; Mangano, C.; Mella, M.; Milanese, C.; Miljkovoc, A. Dicopper(II) MozobilTM: A dinuclear receptor for the pyrophosphate anion in aqueous solution. Supramol. Chem. 2017, 29, 834–845. [Google Scholar] [CrossRef]
- Amendola, V.; Bergamaschi, G.; Buttafava, A.; Fabbrizzi, L.; Monzani, E. Recognition and sensing of nucleoside monophosphates by a dicopper(II) cryptate. J. Am. Chem. Soc. 2010, 132, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, J.; Le Bris, N.; Bencini, A.; Giorgi, C.; Valtancoli, B.; Tripier, R. Glyphosate and ATP binding by mononuclear Zn(II) complexes with non-symmetric ditopic polyamine ligands. Dalton Trans. 2012, 41, 10521–10532. [Google Scholar] [CrossRef] [PubMed]
- Butler, S.J. Ratiometric Detection of Adenosine Triphosphate (ATP) in Water and Real-Time Monitoring of Apyrase Activity with a Tripodal Zinc Complex. Chem. Eur. J. 2014, 20, 15768–15774. [Google Scholar] [CrossRef] [PubMed]
- Carreira-Barral, I.; Rodríguez-Blas, T.; Platas-Iglesias, C.; de Blas, A.; Esteban-Gómez, D. Cooperative Anion Recognition in Copper(II) and Zinc(II) Complexes with a Ditopic Tripodal Ligand Containing a Urea Group. Inorg. Chem. 2014, 53, 2554–2568. [Google Scholar] [CrossRef] [PubMed]
- Carreira-Barral, I.; Mato-Iglesias, M.; de Blas, A.; Platas-Iglesias, C.; Tasker, P.A.; Esteban-Gómez, D. Ditopic receptors containing urea groups for solvent extraction of Cu(II) salts. Dalton Trans. 2017, 46, 3192–3206. [Google Scholar] [CrossRef] [PubMed]
- Djanashvili, K.; Frullano, L.; Peters, J.A. Molecular recognition of sialic acid end groups by phenylboronates. Chem. Eur. J. 2005, 11, 4010–4018. [Google Scholar] [CrossRef] [PubMed]
- Regueiro-Figueroa, M.; Djanashvili, K.; Esteban-Gómez, D.; de Blas, A.; Platas-Iglesias, C.; Rodríguez-Blas, T. Towards Selective Recognition of Sialic Acid Through Simultaneous Binding to Its cis-Diol and Carboxylate Functions. Eur. J. Org. Chem. 2010, 3237–3248. [Google Scholar] [CrossRef]
- Regueiro-Figueroa, M.; Djanashvili, K.; Esteban-Gómez, D.; Chauvin, T.; Tóth, É.; de Blas, A.; Rodríguez-Blas, T.; Platas-Iglesias, C. Molecular Recognition of Sialic Acid by Lanthanide(III) Complexes through Cooperative Two-Site Binding. Inorg. Chem. 2010, 49, 4212–4223. [Google Scholar] [CrossRef] [PubMed]
- Tandon, S.S.; McKee, V.J. Lead(II) and barium(II) complexes of a potentially octadentate macrocyclic ligand (H4L), capable of providing endogenous alkoxy and phenoxy bridges. The X-ray crystal structures of [Pb(H4L)][ClO4]2, [Pb(H2L)][ClO4]2, and [(H6L)(H2O)2][ClO4]2. J. Chem. Soc. Dalton Trans. 1989, 19–24. [Google Scholar] [CrossRef]
- Habata, Y.; Akabori, S. Molecular structure of novel polymer-like complexes of armed-azacrown ethers with alkali-metal cations. J. Chem. Soc. Dalton Trans. 1996, 3871–3882. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents fot characterization of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Halcrow, M.A. Jahn-Teller distortions in transition metal compounds, and their importance in functional molecular materials. Coord. Chem. Rev. 2013, 42, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Hay, B.P.; Dixon, D.A.; Bryan, J.C.; Moyer, B.A. Crystallographic evidence for oxygen acceptor directionality in oxyanion hydrogen bonds. J. Am. Chem. Soc. 2002, 124, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Ikotun, O.F.; Ouellette, W.; Lloret, F.; Kruger, P.E.; Julve, M.; Doyle, R.P. Synthesis, structural, thermal and magnetic characterization of a pyrophosphate-bridged cobalt(II) complex. Eur. J. Inorg. Chem. 2008, 2691–2697. [Google Scholar] [CrossRef]
- Sartoris, R.P.; Nascimento, O.R.; Santana, R.C.; Perec, M.; Baggio, R.F.; Calvo, R. Structure and Magnetism of a binuclear Cu(II) pyrophosphate: Transition to a 3D magnetic behaviour studied by single cristal EPR. Dalton Trans. 2015, 44, 4732–4743. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, J.; Lah, M.S.; Chin, J.; Hong, J.-I. High affinity pyrophosphate receptor by a synergistic effect between metal coordination and hydrogen bonding in water. Org. Lett. 2007, 9, 3729–3731. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Lee, D.H.; Hong, J.-I.; Yoon, J. Chemosensors for pyrophosphate. Acc. Chem. Res. 2009, 42, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Evans, R.; Deng, Z.; Rogerson, A.K.; McLachlan, A.S.; Richards, J.J.; Nilsson, M.; Morris, G.A. Quantitative Interpretation of Diffusion-Ordered NMR Spectra: Can We Rationalize Small Molecule Diffusion Coefficients? Angew. Chem. Int. Ed. 2013, 52, 3199–3202. [Google Scholar] [CrossRef] [PubMed]
- Salvestrini, S.; Di Cerbo, P.; Capasso, S. Kinetics of the chemical degradation of diuron. Chemosphere 2002, 48, 69–73. [Google Scholar] [CrossRef]
- Zambelli, B.; Musiani, F.; Benini, S.; Ciurli, S. Chemistry of Ni2+ in Urease: Sensing, Trafficking, and Catalysis. Acc. Chem. Res. 2011, 44, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Ciurli, S.; Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Mangani, S. Structural properties of the nickel ions in urease: Novel insights into the catalytic and inhibition mechanisms. Coord. Chem. Rev. 1999, 190–192, 331–335. [Google Scholar] [CrossRef]
- Zimmer, M. Molecular Mechanics Evaluation of the Proposed Mechanisms for the Degradation of Urea by Urease. J. Biomol. Struct. Dyn. 2000, 17, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Kistiakowsky, G.B.; Rosenberg, A.J. The Kinetics of Urea Hydrolysis by Urease. J. Am. Chem. Soc. 1952, 74, 5020–5025. [Google Scholar] [CrossRef]
- Krajewska, B. Ureases I. Functional, kinetic and catalytic properties: A review. J. Mol. Catal. B Enzym. 2009, 59, 9–21. [Google Scholar] [CrossRef]
- Dixon, N.E.; Riddles, P.W.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack bean urease (EC 3.5.1.5). V. On the mechanism of action of urease on urea, formamide, acetamide, N-methylurea, and related compounds. Can. J. Biochem. 1980, 58, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.P.; Yunan, Y.; Wolfenden, R. The Burden Borne by Urease. J. Am. Chem. Soc. 2005, 127, 10828–10829. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-Y.; Jeon, Y.-M.; Ryu, H.; Oh, J.-J.; Lim, H.-H.; Kim, M.W. Synthesis and characterization of syn-anti carboxylate-bridged one-dimensional copper(II) complexes with bis(2-pyridylmethyl)amino acids. Polyhedron 2004, 23, 903–911. [Google Scholar] [CrossRef]
- Caneda-Martínez, L.; Valencia, L.; Fernández-Pérez, I.; Regueiro-Figueroa, M.; Angelovski, G.; Brandariz, I.; Esteban-Gómez, D.; Platas-Iglesias, C. Toward inert paramagnetic Ni(II)-based chemical exchange saturation transfer MRI agents. Dalton Trans. 2017, 46, 15095–15106. [Google Scholar] [CrossRef] [PubMed]
- Amendola, V.; Boiocchi, M.; Esteban-Gómez, D.; Fabbrizzi, L.; Monzani, E. Chiral receptors for phosphate ions. Org. Biomol.Chem. 2005, 3, 2632–2639. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Wolsey, W.C. Perchlorate salts, their uses and alternatives. J. Chem. Educ. 1973, 50, A335–A337. [Google Scholar] [CrossRef]
- Shoji, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA Aptamer That Binds the (R)-Isomer of a Thalidomide Derivative with High Enantioselectivity. J. Am. Chem. Soc. 2007, 129, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparition of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. TWINABS—Bruker AXS Scaling for Twinned Crystals—Version 2012/1; Georg-August-Universität Göttingen: Göttingen, Germany, 2012. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX MS-Windows system of programs for solving, refining and analysing single crystal X-ray diffraction data for small molecules. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Beurskens, G.; de Gelder, R.; Garcia-Granda, S.; Gould, R.O.; Smits, J.M.M. The DIRDIF2008 Program System; Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalizated gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401–146405. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Schaefer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Wüllen, C.V. A comparison of density functional methods for the calculation of phosphorous-31 NMR chemical shifts. Phys. Chem. Chem. Phys. 2000, 2, 2137–2144. [Google Scholar] [CrossRef]
- Rezaei-Sameti, M. Ab initio calculations of 31P NMR chemical shielding tensors in alkyl phosphorus compounds and comparison with experimental values. J. Mol. Struct. (Theochem) 2008, 867, 122–124. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [CuL4(SO4)] | [CuL4(H2PPi)] | [{CuL4}2(μ-PPi)] | [{ZnL4}2(μ-PPi)] | |
|---|---|---|---|---|
| M(1)-N(3) | 2.728(3) | 2.670(1) | - | - |
| M(1)-N(4) | 2.044(3) | 2.039(1) | 2.100(2) | 2.340(6) |
| M(1)-N(5) | 2.005(3) | 1.994(1) | 1.990(2) | 2.045(7) |
| M(1)-N(6) | 2.000(2) | 1.991(1) | 1.989(2) | 2.041(6) |
| M(1)-O(4) | 1.960(2) | 1.930(1) | 1.938(1) | 2.021(5) |
| M(1)-O(5) | 2.247(2) | - | - | |
| M(1)-O(8) | - | 2.263(1) | 2.172(2) | 1.949(5) |
| d(D-H)/Å | d(H⋯A)/Å | d(D⋯A)/Å | D-H⋯A/° | ||
|---|---|---|---|---|---|
| [CuL4(SO4)] | N(2)-H(2N)⋯O(6) | 0.82(3) | 1.97(3) | 2.768(3) | 161(3) |
| [{CuL4}2(μ-PPi)] | N(2)-H(2N)⋯O(10) | 0.79(2) | 2.04(3) | 2.830(3) | 175(2) |
| N(3)-H(3N)⋯O(8) | 0.74(3) | 2.08(3) | 2.797(2) | 161(3) | |
| N(8)-H(8N)⋯O(6) | 0.79(2) | 1.98(3) | 2.765(2) | 173(2) | |
| N(9)-H(9N)⋯O(5) | 0.82(3) | 1.94(3) | 2.751(2) | 169(2) | |
| [{ZnL4}2(μ-PPi)] | N(2)-H(2N)⋯O(10) | 0.73(9) | 2.12(9) | 2.850(9) | 172(10) |
| N(3)-H(3N)⋯O(8) | 0.76(9) | 2.13(9) | 2.880(9) | 168(10) | |
| N(8)-H(8N)⋯O(6) | 0.82(9) | 2.05(9) | 2.870(8) | 178(9) | |
| N(9)-H(9N)⋯O(5) | 0.75(9) | 2.15(9) | 2.876(9) | 163(10) |
| Phosphate | AMP | CMP | UMP | PPi | ADP | ATP | ||
|---|---|---|---|---|---|---|---|---|
| [CuL3]2+ | Log K11 | 4.20(1) | 4.35(1) | 3.59(1) | 4.02(1) | 4.82(4) | 4.02(1) | 4.28(1) |
| Log K21 | - | - | - | - | 4.14(5) | 2.42(4) | 3.00(4) | |
| pA | 5.16 | 5.31 | 4.56 | 4.98 | 7.77 | 5.44 | 6.15 | |
| [CuL4]2+ | Log K11 | 3.44(1) | 3.80(1) | 3.31(1) | 2 | 2 | 6.83(9) | 6.26(5) |
| Log K21 | 1.72(7) | - | - | 4.8(1) | 4.32(6) | |||
| pA | 4.56 | 4.76 | 4.29 | 10.44 | 9.39 | |||
| [CuL5]2+ | Log K11 | 3.45(3) | 3.31(3) | 3.51(1) | 3.00(1) | 6.1(3) | 4.89(2) | 4.61(1) |
| Log K21 | 3.67(7) | - | - | - | - | 3.81(2) | 3.41(3) | |
| pA | 5.94 | 4.29 | 4.48 | 4.00 | 11.01 | 7.52 | 6.85 |
| Species | δ (Experimental) | δ (Calculated) Deprotonated Form | δ (Calculated) Doubly Protonated Form |
|---|---|---|---|
| [{ZnL4}2(μ-PPi)] | −5.00 | −5.90 | −10.37 |
| [ZnL4(H2PPi)] | −7.75 | −72.15 | −6.74 |
| [CuL4(SO4)] | [CuL4(H2PPi)] | [{CuL4}2(μ-PPi)] | [{ZnL4}2(μ-PPi)] | |
|---|---|---|---|---|
| formula | C21H22CuN6O7S | C23H29CuN7O11P2 | C42H44Cu2N12O13P2 | C42H56N12O19P2Zn |
| MW | 566.04 | 705.01 | 1113.91 | 1225.66 |
| crystal system | orthorhombic | triclinic | triclinic | monoclinic |
| space group | Pbca | P −1 | P −1 | P 21/n |
| T/K | 100(2) | 100(2) | 100(2) | 100(2) |
| a/Å | 19.636(3) | 7.606(2) | 13.5195(9) | 10.2371(6) |
| b/Å | 8.913(1) | 11.984(3) | 13.6036(9) | 17.6978(10) |
| c/Å | 25.692(3) | 17.360(4) | 14.5281(10) | 28.5263(16) |
| α/deg | 90 | 106.18(1) | 76.346(4) | 90 |
| β/deg | 90 | 95.12(1) | 79.212(5) | 95.104(3) |
| γ/deg | 90 | 105.26(1) | 76.738(4) | 90 |
| V/Å3 | 4496.30(10) | 1443.5(6) | 2502.0(3) | 5147.7(5) |
| F(000) | 2328 | 726 | 1144 | 2536 |
| Z | 8 | 2 | 2 | 4 |
| λ, Å (Mo Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Dcalc/g·cm−3 | 1.672 | 1.622 | 1.479 | 1.581 |
| μ/mm−1 | 1.123 | 0.939 | 0.987 | 1.081 |
| θ range/deg | 2.969–25.149 | 1.855–30.706 | 1.457–26.389 | 1.356–26.391 |
| Rint | 0.0943 | 0.0424 | 0.0391 | 0.1223 c |
| reflns obsd | 4016 | 8880 | 10203 | 13103 |
| GOF on F2 | 1.051 | 1.013 | 1.023 | 1.011 |
| R1 a | 0.038 | 0.0300 | 0.0320 | 0.0768 |
| wR2 (all data) b | 0.0980 | 0.0746 | 0.0765 | 0.1789 |
| Largest diff. peak and hole/e Å−3 | 0.464 and −0.533 | 0.532 and −0.366 | 0.397 and −0.434 | 0.959 and −1.946 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreira-Barral, I.; Fernández-Pérez, I.; Mato-Iglesias, M.; De Blas, A.; Platas-Iglesias, C.; Esteban-Gómez, D. Recognition of AMP, ADP and ATP through Cooperative Binding by Cu(II) and Zn(II) Complexes Containing Urea and/or Phenylboronic—Acid Moieties. Molecules 2018, 23, 479. https://doi.org/10.3390/molecules23020479
Carreira-Barral I, Fernández-Pérez I, Mato-Iglesias M, De Blas A, Platas-Iglesias C, Esteban-Gómez D. Recognition of AMP, ADP and ATP through Cooperative Binding by Cu(II) and Zn(II) Complexes Containing Urea and/or Phenylboronic—Acid Moieties. Molecules. 2018; 23(2):479. https://doi.org/10.3390/molecules23020479
Chicago/Turabian StyleCarreira-Barral, Israel, Isabel Fernández-Pérez, Marta Mato-Iglesias, Andrés De Blas, Carlos Platas-Iglesias, and David Esteban-Gómez. 2018. "Recognition of AMP, ADP and ATP through Cooperative Binding by Cu(II) and Zn(II) Complexes Containing Urea and/or Phenylboronic—Acid Moieties" Molecules 23, no. 2: 479. https://doi.org/10.3390/molecules23020479
APA StyleCarreira-Barral, I., Fernández-Pérez, I., Mato-Iglesias, M., De Blas, A., Platas-Iglesias, C., & Esteban-Gómez, D. (2018). Recognition of AMP, ADP and ATP through Cooperative Binding by Cu(II) and Zn(II) Complexes Containing Urea and/or Phenylboronic—Acid Moieties. Molecules, 23(2), 479. https://doi.org/10.3390/molecules23020479