Bacillus Cellulase Molecular Cloning, Expression, and Surface Display on the Outer Membrane of Escherichia coli



Abstract
:1. Introduction
2. Results and Discussion
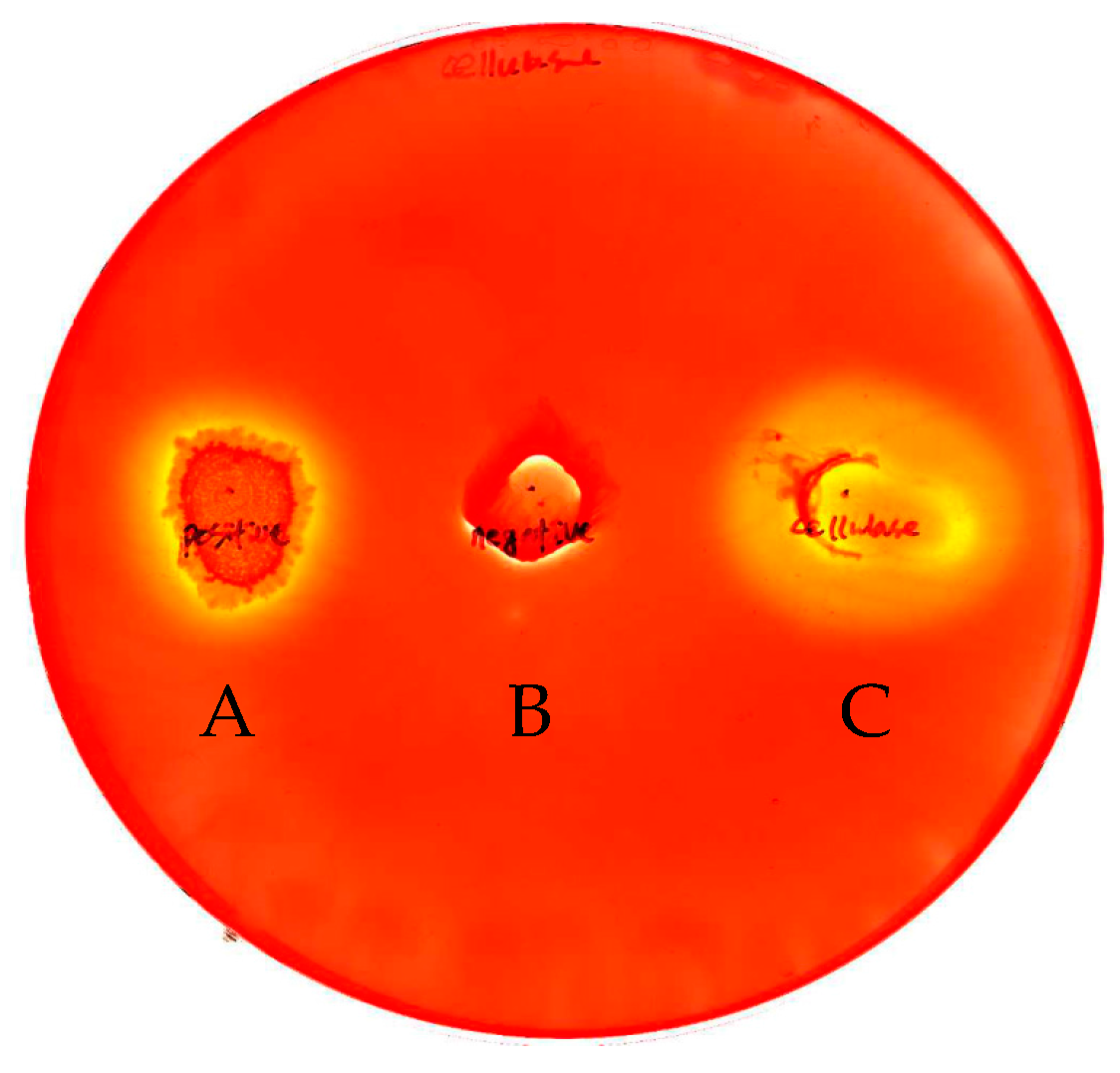
2.1. Cloning and Expression of the INP-Cellulase Gene
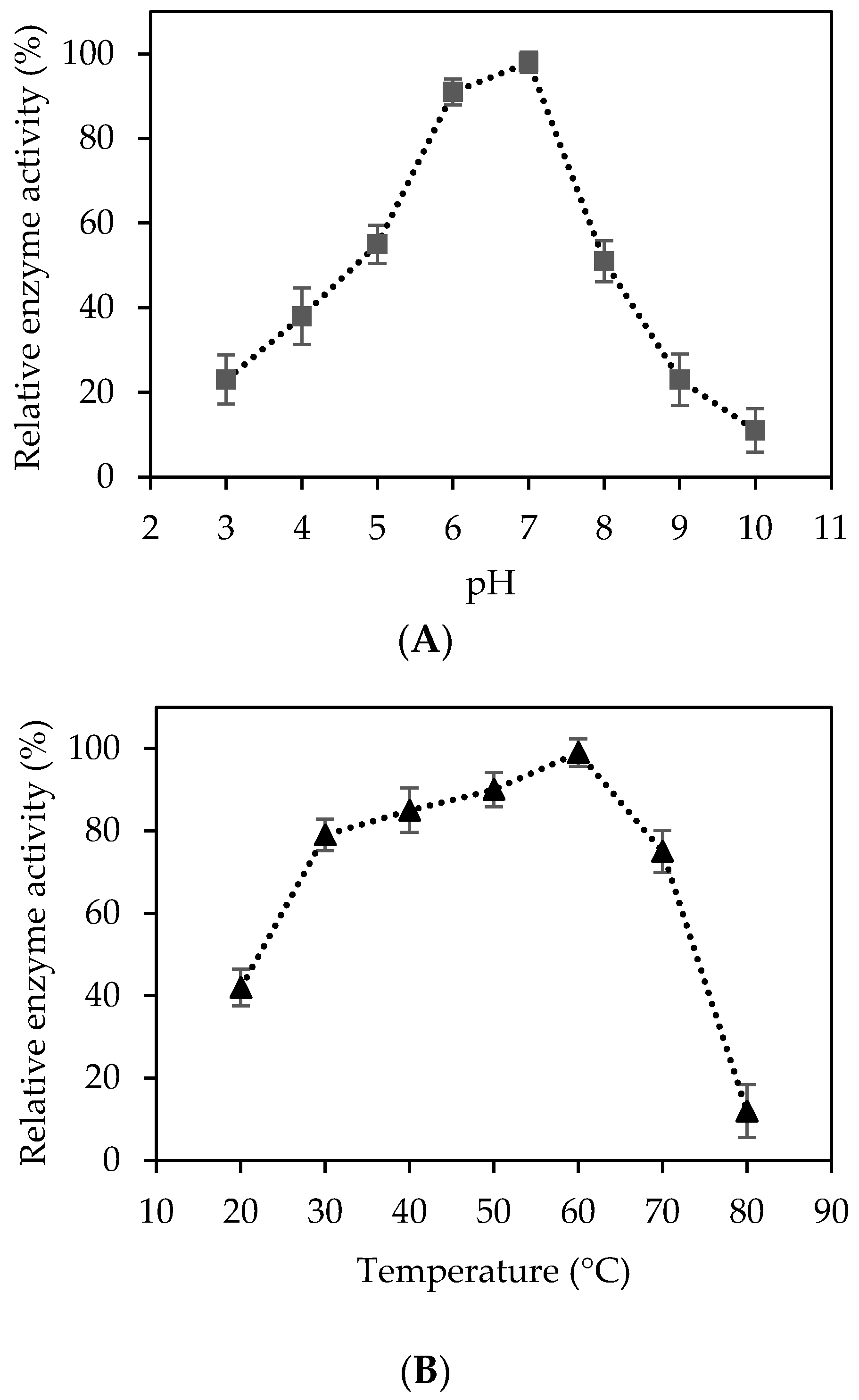
2.2. Characterization of Recombinant Cellulase
3. Materials and Methods
3.1. Materials
3.2. Bacterial Strains, Plasmids, and Culture Conditions
3.3. Plasmid Construction
3.4. Cloning and Sequence Analysis
3.5. Cell Fractionation
3.6. Analytical Methods
3.7. Enzyme Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Feng, Y.; Zhang, M.; Mujumdar, A.S.; Gao, Z. Recent research process of fermented plant extract: A review. Trends Food Sci. Technol. 2017, 65, 40–48. [Google Scholar] [CrossRef]
- Hasunuma, T.; Okazaki, F.; Okai, N.; Hara, K.Y.; Ishii, J.; Kondo, A. A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology. Bioresour. Technol. 2013, 135, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Ku, S. Beneficial Effects of Monascus sp. KCCM 10093 Pigments and Derivatives: A Mini Review. Molecules 2018, 23, 98. [Google Scholar] [CrossRef] [PubMed]
- Ku, S. Finding and Producing Probiotic Glycosylases for the Biocatalysis of Ginsenosides: A Mini Review. Molecules 2016, 21, 645. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Park, M.S.; Ji, G.E.; You, H.J. Review on bifidobacterium bifidum bgn4: Functionality and nutraceutical applications as a probiotic microorganism. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Vibbert, H.B.; Ku, S.; Li, X.; Liu, X.; Ximenes, E.; Kreke, T.; Ladisch, M.R.; Deering, A.J.; Gehring, A.G. Accelerating sample preparation through enzyme-assisted microfiltration of Salmonella in chicken extract. Biotechnol. Prog. 2015, 31, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ku, S.; Park, M.S.; Li, Z.; Ji, G.E. Acceleration of Aglycone Isoflavone and γ-Aminobutyric Acid Production from Doenjang Using Whole-Cell Biocatalysis Accompanied by Protease Treatment. J. Microbial. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; You, H.J.; Park, M.S.; Ji, G.E. Whole-cell biocatalysis for producing ginsenoside rd from Rb1 using Lactobacillus rhamnosus GG. J. Microbiol. Biotechnol. 2016, 26, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Juturu, V.; Wu, J.C. Microbial cellulases: Engineering, production and applications. Renew. Sustain. Energy Rev. 2014, 33, 188–203. [Google Scholar] [CrossRef]
- Kuhad, R.C.; Gupta, R.; Singh, A. Microbial cellulases and their industrial applications. Enzyme Res. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, R.K.; Singhania, R.R.; Pandey, A. Microbial cellulases—Production, applications and challenges. J. Sci. Ind. Res. (India) 2005, 64, 832–844. [Google Scholar]
- Phitsuwan, P.; Laohakunjit, N.; Kerdchoechuen, O.; Kyu, K.L.; Ratanakhanokchai, K. Present and potential applications of cellulases in agriculture, biotechnology, and bioenergy. Folia Microbiol. (Praha) 2013, 58, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Foreman, P.K.; Brown, D.; Dankmeyer, L.; Dean, R.; Diener, S.; Dunn-Coleman, N.S.; Goedegebuur, F.; Houfek, T.D.; England, G.J.; Kelley, A.S.; et al. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J. Biol. Chem. 2003, 278, 31988–31997. [Google Scholar] [CrossRef] [PubMed]
- Segato, F.; Damásio, A.R.L.; de Lucas, R.C.; Squina, F.M.; Prade, R.A. Genomics Review of holocellulose deconstruction by aspergilli. Microbiol. Mol. Biol. Rev. 2014, 78, 588–613. [Google Scholar] [CrossRef] [PubMed]
- Kozloff, L.M.; Turner, M.A.; Arellano, F. Formation of bacterial membrane ice-nucleoting lipoglycoprotein complexes. J. Bacteriol. 1991, 173, 6528–6536. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.; Ståhl, S.; Nguyen, T.N.; Bächi, T.; Robert, A.; Binz, H.; Sjölander, A.; Uhlén, M. Expression of recombinant proteins on the surface of the coagulase-negative bacterium Staphylococcus xylosus. J. Bacteriol. 1992, 174, 4239–4245. [Google Scholar] [CrossRef] [PubMed]
- Sleytr, U.B.; Sára, M. Bacterial and archaeal S-layer proteins: Structure-function relationships and their biotechnological applications. Trends Biotechnol. 1997, 15, 20–26. [Google Scholar] [CrossRef]
- Kojima, M.; Akahoshi, T.; Okamoto, K.; Yanase, H. Expression and surface display of Cellulomonas endoglucanase in the ethanologenic bacterium Zymobacter palmae. Appl. Microbiol. Biotechnol. 2012, 96, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, Q.; Chen, J.; He, Y.; Wang, J.; Zhang, H.; Yu, Z.; Li, L. Molecular characterization of an ice nucleation protein variant (InaQ) from Pseudomonas syringae and the analysis of its transmembrane transport activity in Escherichia coli. Int. J. Biol. Sci. 2012, 8, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.C.; Park, J.H.; Park, S.H.; Lebeault, J.M.; Pan, J.G. Expression of carboxymethylcellulase on the surface of Escherichia coli using Pseudomonas syringae ice nucleation protein. Enzyme Microb. Technol. 1998, 22, 348–354. [Google Scholar] [CrossRef]
- Jung, H.-C.; Lebeault, J.-M.; Pan, J.-G. Surface display of Zymomonas mobilis levansucrase by using the ice-nucleation protein of Pseudomonas syringae. Nat. Biotechnol. 1998, 16, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Van Bloois, E.; Winter, R.T.; Kolmar, H.; Fraaije, M.W. Decorating microbes: Surface display of proteins on Escherichia coli. Trends Biotechnol. 2011, 29, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, K.D. Protein engineering by cell-surface display. Curr. Opin. Biotechnol. 2001, 12, 395–399. [Google Scholar] [CrossRef]
- Jose, J. Autodisplay: Efficient bacterial surface display of recombinant proteins. Appl. Microbiol. Biotechnol. 2006, 69, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Campbell, R.; Iverson, B.L.; Georgiou, G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. USA 1993, 90, 10444–10448. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, A.P.; Kornacker, M.G. Secretion of the cell surface lipoprotein pullulanase in Escherichia coli. J. Biol. Chem. 1991, 266, 13640–13645. [Google Scholar] [PubMed]
- Gao, G.; Mao, R.Q.; Xiao, Y.; Zhou, J.; Liu, Y.H.; Li, G. Efficient yeast cell-surface display of an endoglucanase of Aspergillus flavus and functional characterization of the whole-cell enzyme. World J. Microbiol. Biotechnol. 2017, 33, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Washida, M.; Takahashi, S.; Ueda, M.; Tanaka, A. Spacer-mediated display of active lipase on the yeast cell surface. Appl. Microbiol. Biotechnol. 2001, 56, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamada, R.; Ogino, C.; Kondo, A. Recent developments in yeast cell surface display toward extended applications in biotechnology. Appl. Microbiol. Biotechnol. 2012, 95, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, M.; Mulchandani, A.; Chen, W. Cell surface display of organophosphorus hydrolase using ice nucleation protein. Biotechnol. Prog. 2001, 17, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Kotrba, P.; Dolecková, L.; De Lorenzo, V.; Kotrba, P.; Dolec, L. Enhanced bioaccumulation of heavy metal ions by bacterial cells due to surface display of short metal binding peptides enhanced bioaccumulation of heavy metal ions by bacterial cells due to surface display of short metal binding peptides. Appl. Environ. Microbiol. 1999, 65, 1092–1098. [Google Scholar] [PubMed]
- Mejàre, M.; Ljung, S.; Bülow, L. Selection of cadmium specific hexapeptides and their expression as OmpA fusion proteins in Escherichia coli. Protein Eng. 1998, 11, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Choi, J.H.; Xu, Z. Microbial cell-surface display. Trends Biotechnol. 2003, 21, 45–52. [Google Scholar] [CrossRef]
- Löfblom, J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011, 6, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Stentebjerg-Olesen, B.; Pallesen, L.; Jensen, L.B.; Christiansen, G.; Klemm, P. Authentic display of a cholera toxin epitope by chimeric type 1 fimbriae: Effects of insert position and host background. Microbiology 1997, 143, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Chen, G.; Olsen, M.J.; Iverson, B.L.; Georgiou, G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998, 11, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Olsen, M.J.; Iverson, B.L.; Georgiou, G. Development of an optimized expression system for the screening of antibody libraries displayed on the Escherichia coli surface. Protein Eng. 1999, 12, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Chen, G.; Iverson, B.L.; Georgiou, G. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain Fv antibodies. Proc. Natl. Acad. Sci. USA 2000, 97, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, P.S.; Iverson, B.L.; Georgiou, G. Flow cytometric screening of cell-based libraries. J. Immunol. Methods 2000, 243, 211–227. [Google Scholar] [CrossRef]
- Shi, H.; Wen Su, W. Display of green fluorescent protein on Escherichia coli cell surface. Enzyme Microb. Technol. 2001, 28, 25–34. [Google Scholar] [CrossRef]
- Sun, F.; Pang, X.; Xie, T.; Zhai, Y.; Wang, G.; Sun, F. BrkAutoDisplay: Functional display of multiple exogenous proteins on the surface of Escherichia coli by using BrkA autotransporter. Microb. Cell Fact. 2015, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Margaritis, A.; Bassi, A.S. Principles and biotechnological applications of bacterial ice nucleation. Crit. Rev. Biotechnol. 1991, 11, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Saito, S.; Ohmine, I.; Mn, D.; Am, J.; Macroscopic, R.; Quantum, R.; Quantum, D.; The, G.R.; Hydrogen, P.G. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nature 2002, 416, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Nemecek-Marshall, M.; LaDuca, R.; Fall, R. High-level expression of ice nuclei in a Pseudomonas syringae strain is induced by nutrient limitation and low temperature. J. Bacteriol. 1993, 175, 4062–4070. [Google Scholar] [CrossRef] [PubMed]
- Maki, L.R.; Galyan, E.L.; Chang-Chien, M.M.; Caldwell, D.R. Ice nucleation induced by pseudomonas syringae. Appl. Microbiol. 1974, 28, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Drainas, C.; Vartholomatos, G.; Panopoulos, N.J.; Ice, T.; Gene, N. The ice nucleation gene from pseudomonas syringae as a sensitive gene reporter for promoter analysis in zymomonas mobilis the ice nucleation gene from pseudomonas syringae as a sensitive gene reporter for promoter analysis in zymomonas mobilis. Appl. Environ. Microbiol. 1995, 61, 273–277. [Google Scholar] [PubMed]
- Li, Q.; Yu, Z.; Shao, X.; He, J.; Li, L. Improved phosphate biosorption by bacterial surface display of phosphate-binding protein utilizing ice nucleation protein. FEMS Microbiol. Lett. 2009, 299, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Lee, H.; Byun, D.; Choi, H.; Shim, J.H. Molecular cloning, characterization, and application of a novel thermostable α-glucosidase from the hyperthermophilic archaeon Pyrobaculum aerophilum strain IM2. Food Sci. Biotechnol. 2015, 24, 175–182. [Google Scholar] [CrossRef]
- Wolber, P.; Deininger, C.; Southworth, M.; Vandekerckhove, J.; van Montagu, M.; Warren, G. Identification and purification of a bacterial ice-nucleation protein. Proc. Natl. Acad. Sci. USA 1986, 83, 7256–7260. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.W.; Ramaiya, P.; Nelson, B.A.; Brody-Karpin, S.D.; Zaretsky, E.J.; Tang, M.; Lopez de, L.A.; Xiang, H.; Gusti, V.; Clausen, I.G.; et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 2004, 5, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, N.; Okamoto, T.; Nakamura, K. Expression of a cellulase gene in Zymomonas mobilis. J. Biotechnol. 1988, 7, 167–177. [Google Scholar] [CrossRef]
- Lejeune, A.; Eveleigh, D.E.; Colson, C. Expression of an endoglucanase gene of Pseudomonas fluorescens var. cellulosa in Zymomonas mobilis. FEMS Microbiol. Lett. 1988, 49, 363–366. [Google Scholar] [CrossRef]
- Thirumalai Vasan, P.; Sobana Piriya, P.; Immanual Gilwax Prabhu, D.; John Vennison, S. Cellulosic ethanol production by Zymomonas mobilis harboring an endoglucanase gene from Enterobacter cloacae. Bioresour. Technol. 2011, 102, 2585–2589. [Google Scholar] [CrossRef] [PubMed]
- Borgi, M.A.; Boudebbouze, S.; Aghajari, N.; Szukala, F.; Pons, N.; Maguin, E.; Rhimi, M. The attractive recombinant phytase from Bacillus licheniformis: Biochemical and molecular characterization. Appl. Microbiol. Biotechnol. 2014, 98, 5937–5947. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, K.M.; Rooney, A.P.; Li, X.L.; Liu, S.; Hughes, S.R. Purification and characterization of a family 5 endoglucanase from a moderately thermophilic strain of Bacillus licheniformis. Biotechnol. Lett. 2006, 28, 1761–1765. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Wyman, C.E. Strong cellulase inhibition by Mannan polysaccharides in cellulose conversion to sugars. Biotechnol. Bioeng. 2014, 111, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.K. Cellulases and related enzymes in biotechnology. Biotechnol. Adv. 2000, 18, 355–383. [Google Scholar] [CrossRef]
- Divakaran, D.; Chandran, A.; Pratap Chandran, R. Comparative study on production of a-Amylase from Bacillus licheniformis strains. Braz. J. Microbiol. 2011, 42, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.U.; Ashraf, H.; Iqbal, J.; Qadeer, M.A. Production of alpha amylase by Bacillus licheniformis using an economical medium. Bioresour. Technol. 2003, 87, 57–61. [Google Scholar] [CrossRef]
- Mendo, S.; Faustino, N.A.; Sarmento, A.C.; Amado, F.; Moir, A.J.G. Purification and characterization of a new peptide antibiotic produced by a thermotolerant Bacillus licheniformis strain. Biotechnol. Lett. 2004, 26, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Waschkau, B.; Waldeck, J.; Wieland, S.; Eichstädt, R.; Meinhardt, F. Generation of readily transformable Bacillus licheniformis mutants. Appl. Microbiol. Biotechnol. 2008, 78, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-H.; Lee, Y.-S.; Cho, J.-Y.; Ahn, Y.-S.; Moon, J.-H.; Hyun, H.-N.; Cha, G.-S.; Kim, K.-Y. Isolation and characterization of metabolites from Bacillus licheniformis MH48 with antifungal activity against plant pathogens. Microb. Pathog. 2017, 110, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Du, R.; Zhao, D.; Song, G.; Jin, M.; Ping, W. Kinetic study of a β-mannanase from the Bacillus licheniformis HDYM-04 and its decolorization ability of twenty-two structurally different dyes. Springerplus 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Songsiriritthigul, C.; Lapboonrueng, S.; Roytrakul, S.; Haltrich, D.; Yamabhai, M. Crystallization and preliminary crystallographic analysis of β-mannanase from Bacillus licheniformis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Craynest, M.; Jørgensen, S.; Sarvas, M.; Kontinen, V.P. Enhanced secretion of heterologous cyclodextrin glycosyltransferase by a mutant of Bacillus licheniformis defective in the d-alanylation of teichoic acids. Lett. Appl. Microbiol. 2003, 37, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.U.; Aman, A.; Zohra, R.R.; Qader, S.A.U. Immobilization of pectin degrading enzyme from Bacillus licheniformis KIBGE IB-21 using agar-agar as a support. Carbohydr. Polym. 2014, 102, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.U.; Siddique, N.N.; Aman, A.; Nawaz, M.A.; Baloch, A.H.; Qader, S.A.U. Morphological and molecular based identification of pectinase producing Bacillus licheniformis from rotten vegetable. J. Genet. Eng. Biotechnol. 2015, 13, 139–144. [Google Scholar] [CrossRef]
- Li, S.; Yang, X.; Yang, S.; Zhu, M.; Wang, X. Technology Prospecting on Enzymes: Application, Marketing and Engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209017. [Google Scholar] [CrossRef] [PubMed]
- Borgi, M.A.; Boudebbouze, S.; Mkaouar, H.; Maguin, E.; Rhimi, M. Bacillus phytases: Current status and future prospects. Bioengineered 2015, 6, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Tye, A.; Siu, F.; Leung, T.; Lim, B. Molecular cloning and the biochemical characterization of two novel phytases from B. subtilis 168 and B. licheniformis. Appl. Microbiol. Biotechnol. 2002, 59, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, A.; Bando, H.; Kaya, M.; Kato-Murai, M.; Kuroda, K.; Sahara, H.; Hata, Y.; Kondo, A.; Ueda, M. Direct ethanol production from barley β-glucan by sake yeast displaying Aspergillus oryzae β-glucosidase and endoglucanase. J. Biosci. Bioeng. 2008, 105, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Yanase, S.; Hasunuma, T.; Yamada, R.; Tanaka, T.; Ogino, C.; Fukuda, H.; Kondo, A. Direct ethanol production from cellulosic materials at high temperature using the thermotolerant yeast Kluyveromyces marxianus displaying cellulolytic enzymes. Appl. Microbiol. Biotechnol. 2010, 88, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jung, H.; Pan, J. Bacterial Cell Surface Display of an Enzyme Library for Selective Screening of Improved Cellulase Variants Bacterial Cell Surface Display of an Enzyme Library for Selective Screening of Improved Cellulase Variants. Appl. Environ. Microbiol. 2000, 66, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, T.N.; Xu, T.F.; Wang, J.Y.; Wang, C.L.; Zhao, M. Cloning and expression of thermo-alkali-stable laccase of Bacillus licheniformis in Pichia pastoris and its characterization. Bioresour. Technol. 2013, 134, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Jana, M.L.; Sengupta, D.; Naskar, A.K. A note on the estimation of microbial glycosidase activities by dinitrosalicylic acid reagent. Appl. Microbiol. Biotechnol. 2000, 53, 732–735. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Cell Fractionation | Wild Type | Recombinant (INP-Cellulase) | ||
|---|---|---|---|---|
| (mU/mL) | (%) 1 | mU/mL | (%) 1 | |
| Cell Culture Medium (Supernatant) | 17.2 | 14.8 | 468.0 | 47.5 |
| Periplasm | 26.3 | 22.7 | 307.4 | 31.2 |
| Cytoplasmic (Intracellular) | 71.0 | 61.3 | 18.7 | 1.9 |
| Membrane | 1.4 | 1.2 | 191.1 | 19.4 |
| Total Activity | 115.9 | 100 | 985.2 | 100 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Ku, S. Bacillus Cellulase Molecular Cloning, Expression, and Surface Display on the Outer Membrane of Escherichia coli. Molecules 2018, 23, 503. https://doi.org/10.3390/molecules23020503
Kim D, Ku S. Bacillus Cellulase Molecular Cloning, Expression, and Surface Display on the Outer Membrane of Escherichia coli. Molecules. 2018; 23(2):503. https://doi.org/10.3390/molecules23020503
Chicago/Turabian StyleKim, Daehwan, and Seockmo Ku. 2018. "Bacillus Cellulase Molecular Cloning, Expression, and Surface Display on the Outer Membrane of Escherichia coli" Molecules 23, no. 2: 503. https://doi.org/10.3390/molecules23020503
APA StyleKim, D., & Ku, S. (2018). Bacillus Cellulase Molecular Cloning, Expression, and Surface Display on the Outer Membrane of Escherichia coli. Molecules, 23(2), 503. https://doi.org/10.3390/molecules23020503