Screening of Genes Related to Early and Late Flowering in Tree Peony Based on Bulked Segregant RNA Sequencing and Verification by Quantitative Real-Time PCR

Abstract
:
1. Introduction
2. Results
2.1. Sequence Assembly and Annotation of Functional Genes
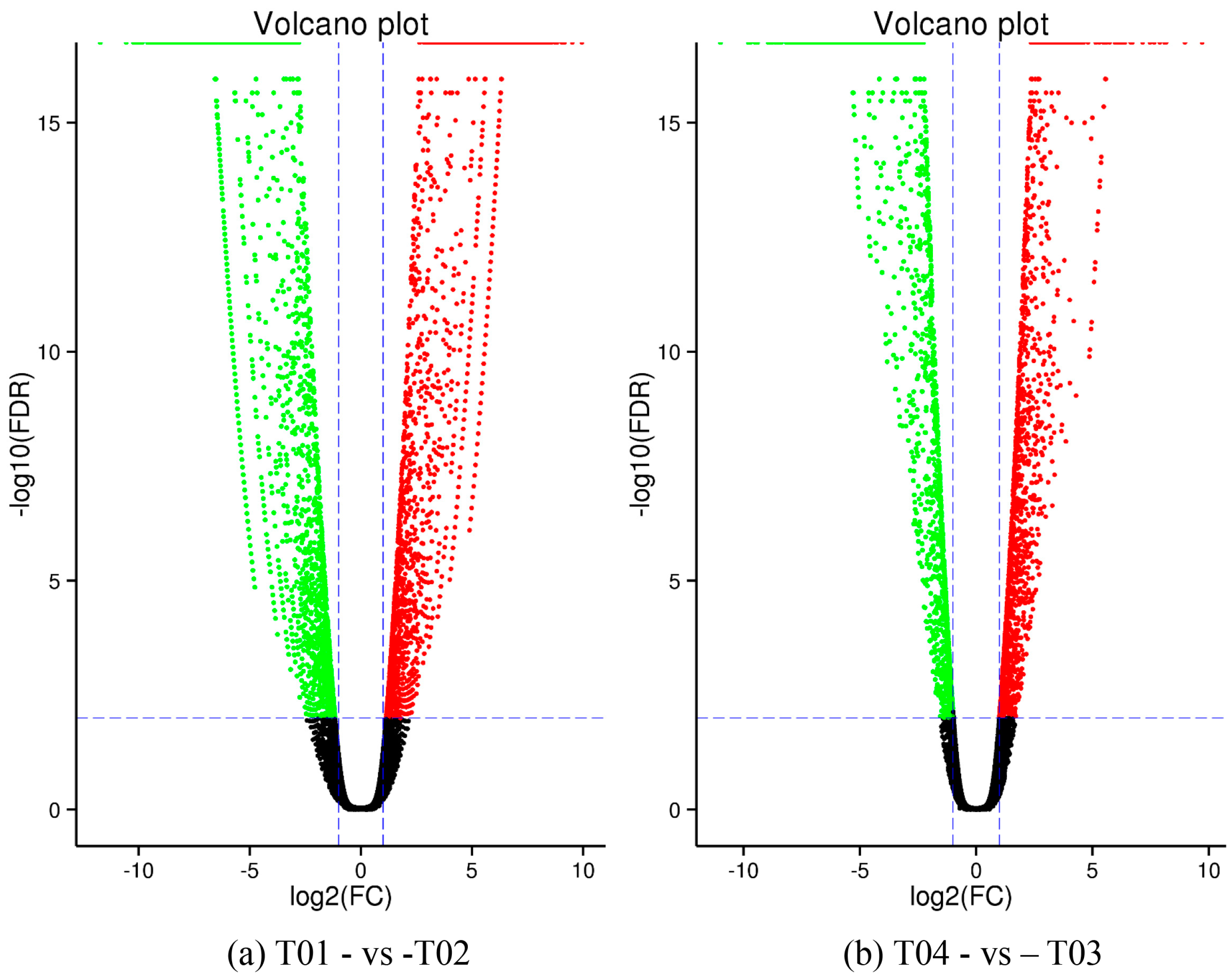
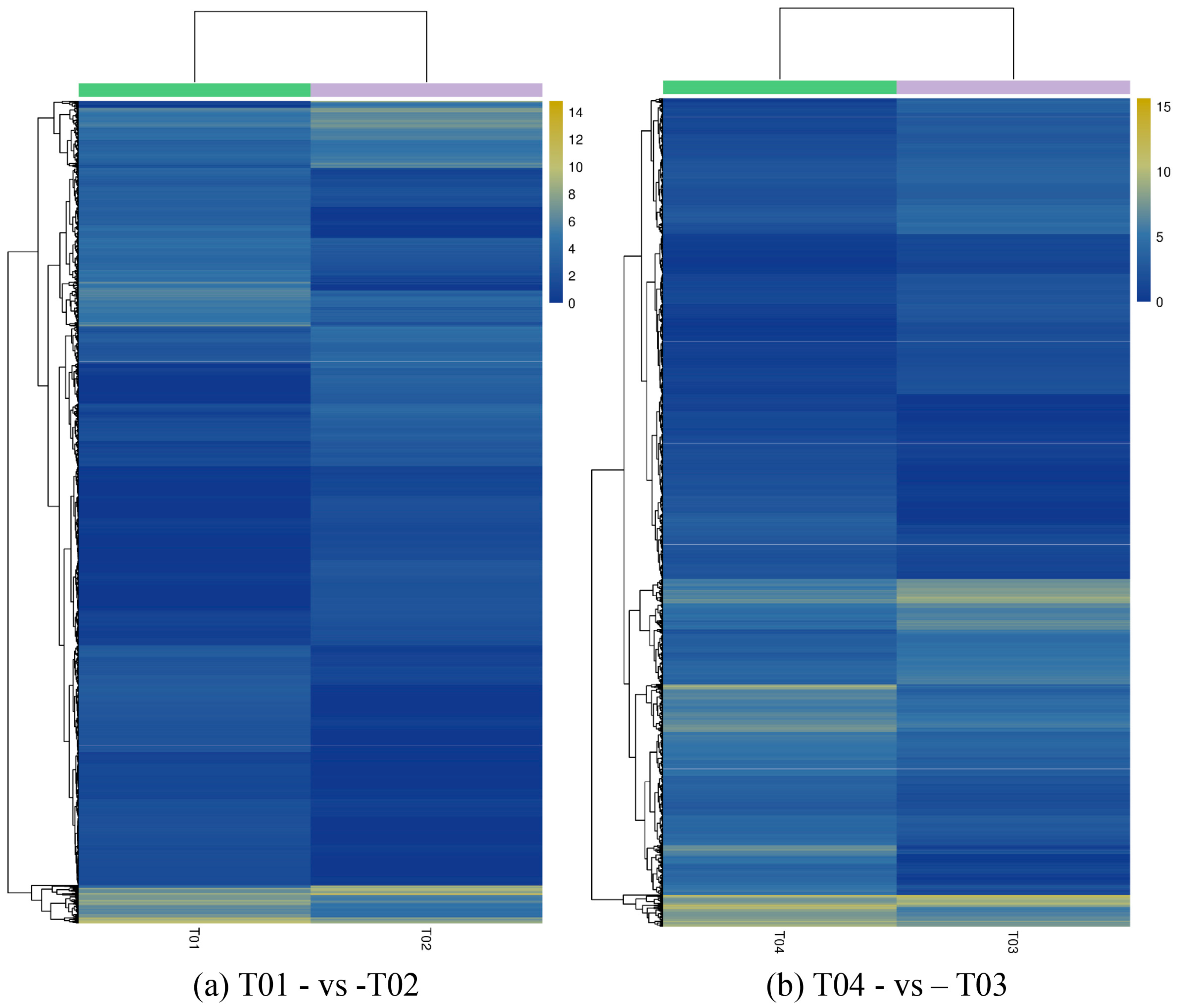
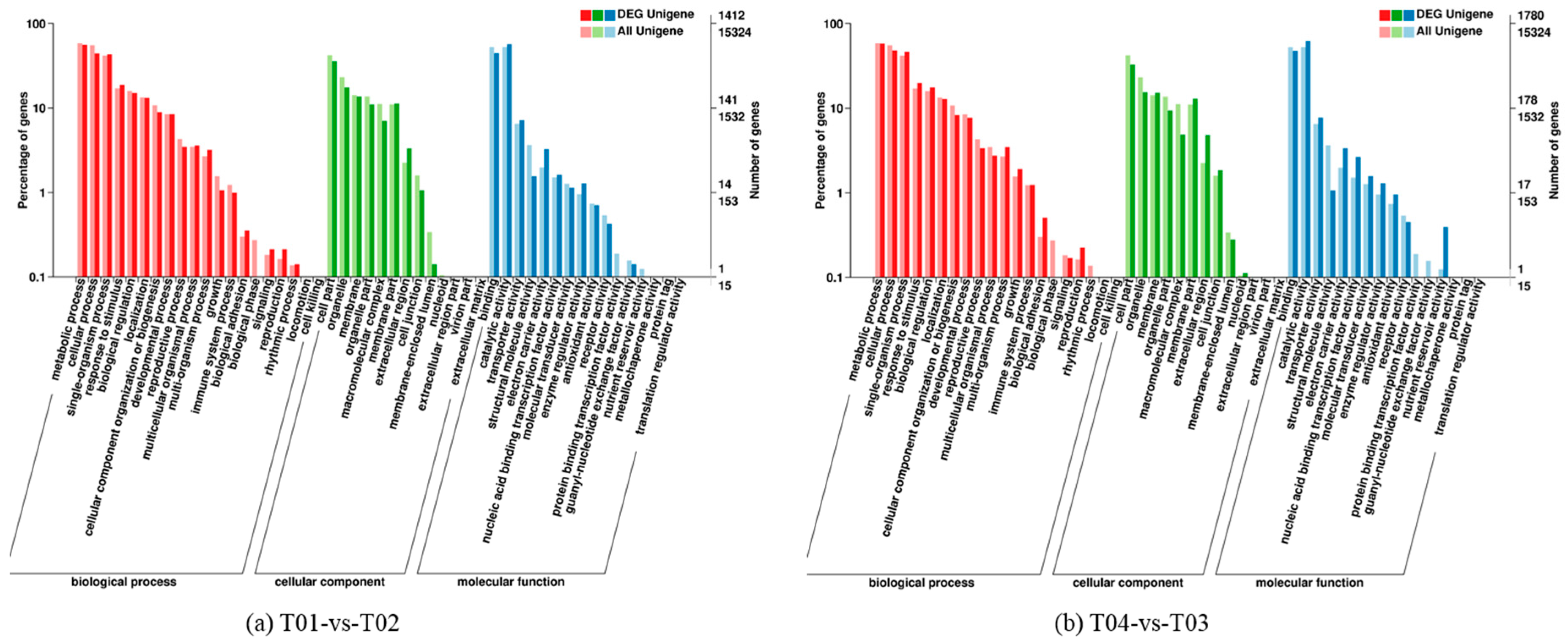
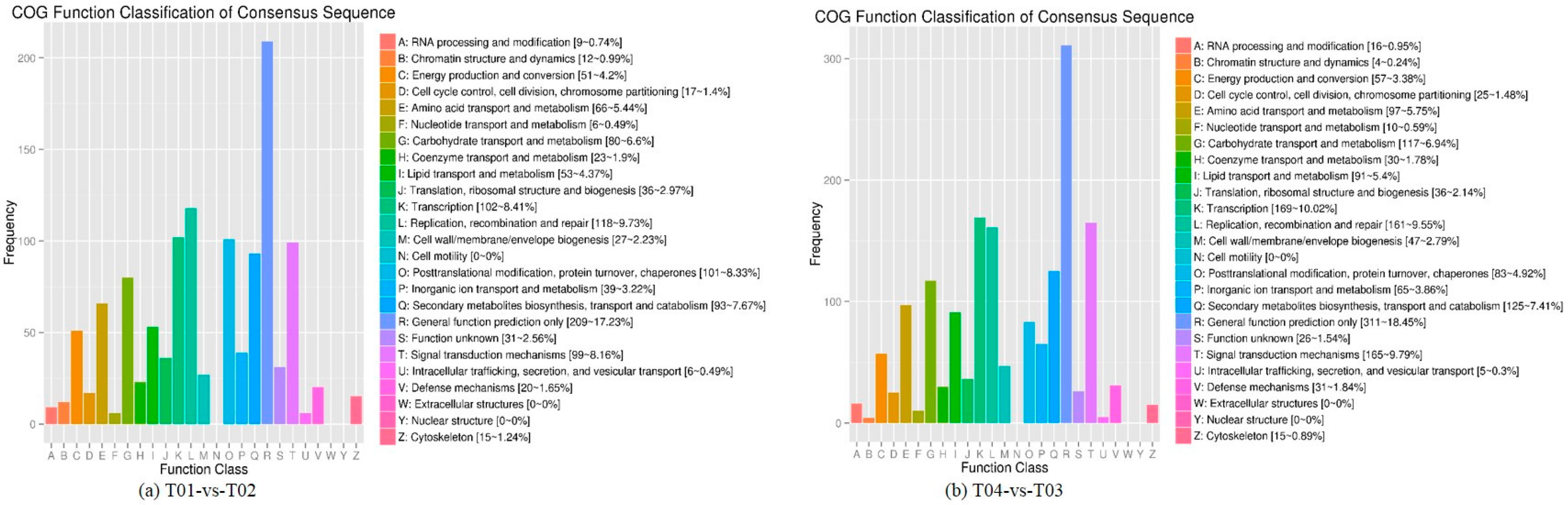
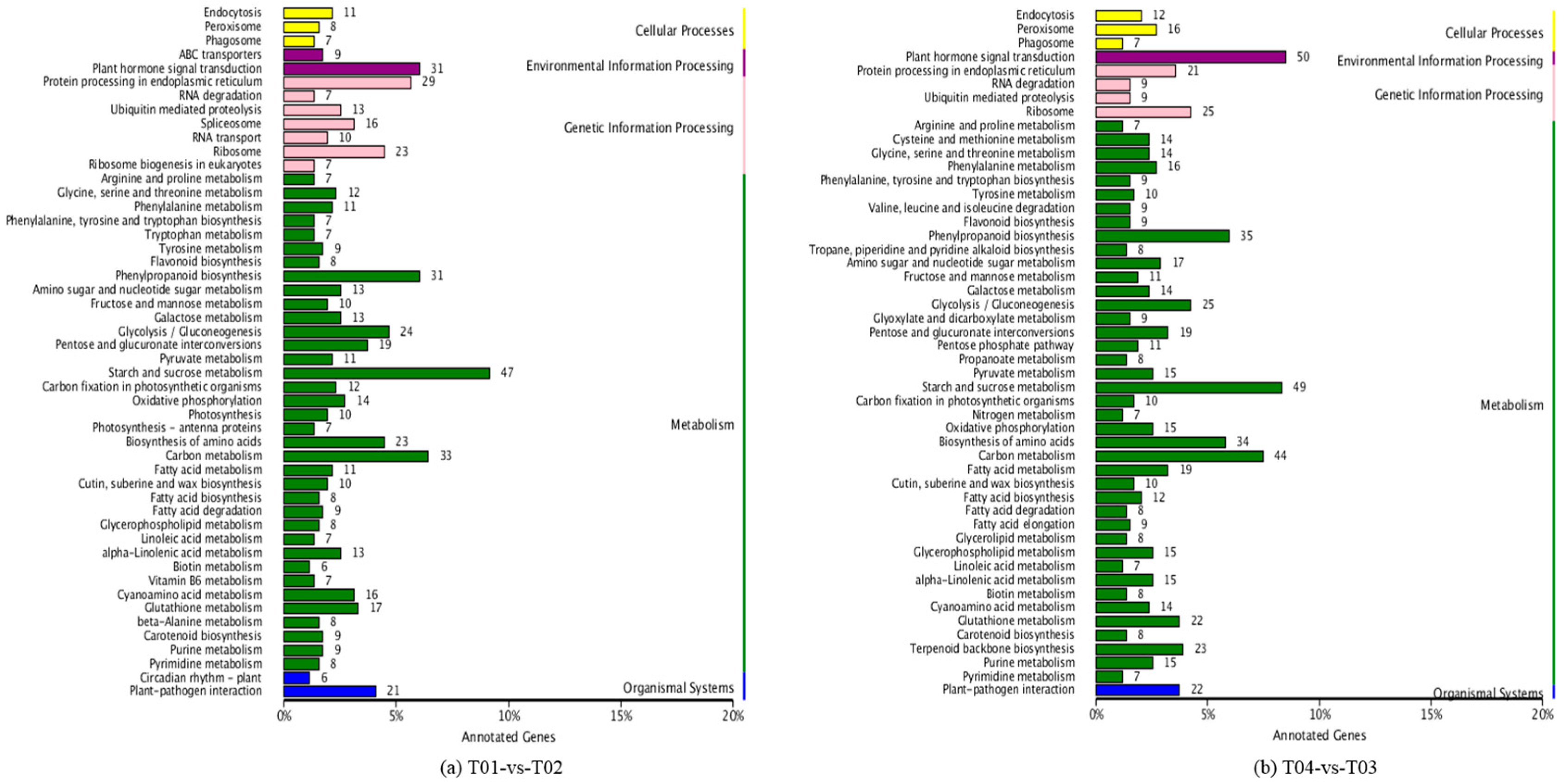
2.2. Analysis of Differentially Expressed Genes (DEGs)
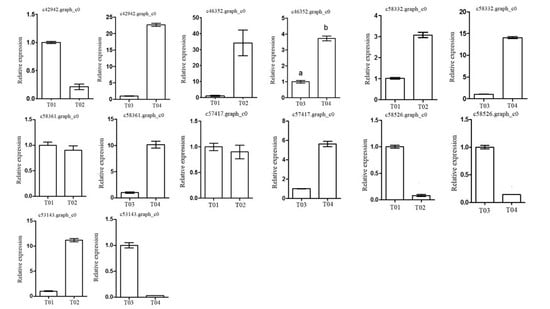
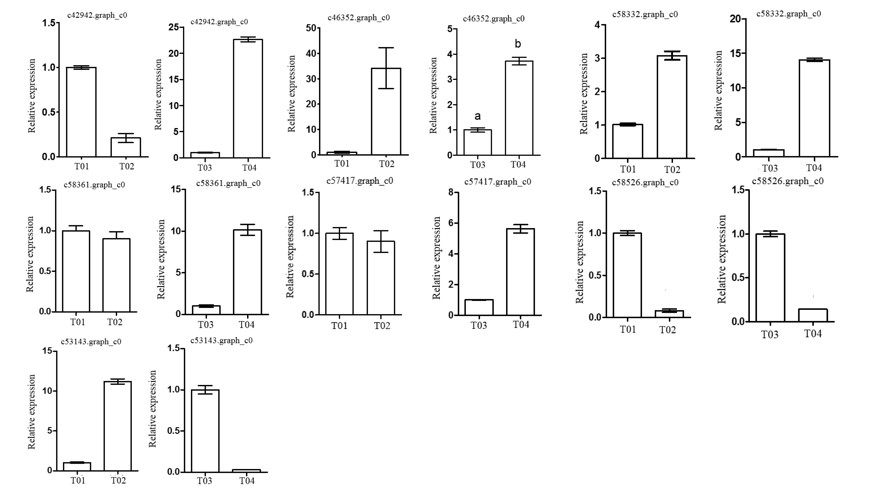
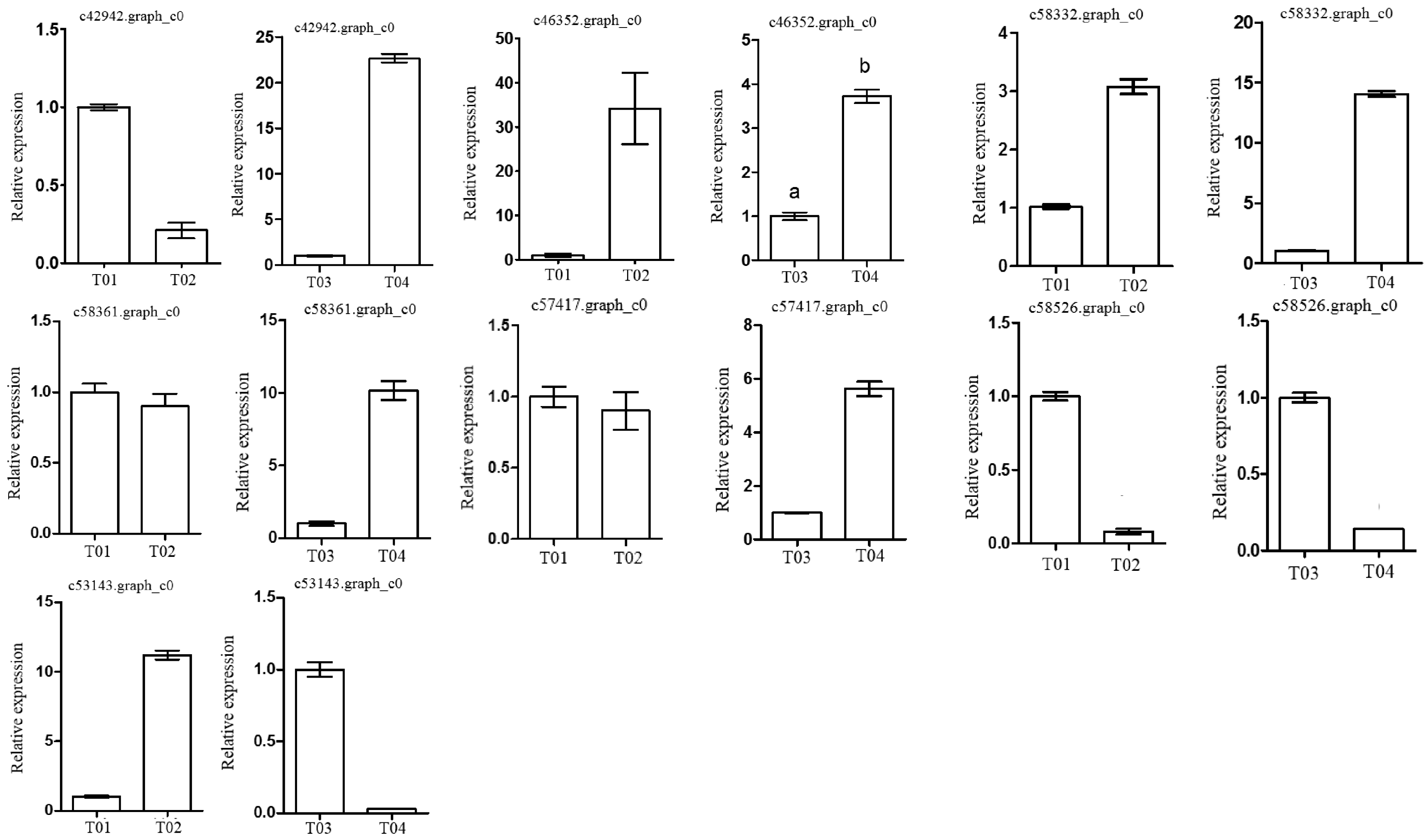
2.3. Verification of Candidate Genes through Quantitative Real-Time PCR (qRT-PCR)
2.4. Simple Sequence Repeats (SSRs) and Single Nucleotide Polymorphisms (SNPs)
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Illumina Sequencing
4.3. De Novo Assembly and Quality Control
4.4. Unigene Functional Annotation and Gene Structure Analysis
4.5. Gene Expression Quantification
4.6. Analysis of Genes with Differential Expression (DEGs)
4.7. BSR-Association Study and Candidate Genes Identification
4.8. Quantitative Real-Time PCR (qRT-PCR) Verification of Candidate Genes
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haw, S. Tree peonies: A review of their history and taxonomy. New Plantsman 2001, 8, 156–171. [Google Scholar]
- Hong, D.Y.; Pan, K.Y. Notes on taxonomy of Paeonia sect Moutan DC. (Paeoniaceae). Acta Phytotax. Sini. 2005, 43, 169–177. [Google Scholar] [CrossRef]
- Li, J.J.; Zhang, X.F.; Zhao, X.Q. Tree Peony of Chinese; Encyclopedia of China Publishing House: Beijing, China, 2011; p. 340. [Google Scholar]
- Rogers, A.; Engstrom, L. Peonies; Timber Press: Portland, OR, USA, 1995; p. 296. [Google Scholar]
- Wang, L.Y. Chinese Tree Peony; China Forestry Publishing House: Beijing, China, 1998; p. 212. [Google Scholar]
- Cheng, F.Y. Advances in the breeding of tree peonies and a cultivar system for the cultivar group. Int. J. Plant Breed. 2007, 2, 89–104. [Google Scholar]
- Koornneef, M.; Alonso-Blanco, C.; Peeters, A.J.M.; Soppe, W. Genetic control of flowering time in Arabiciopsis. Annu. Rev. Plant Biol. 1998, 49, 345–370. [Google Scholar] [CrossRef] [PubMed]
- Reeves, P.H.; Coupland, G. Analysis of flowering time control in Arabidopsis by comparison of double and triple mutants. Plant Physiol. 2001, 126, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Sung, Z.R.; Chen, L.J.; Moon, Y.H.; Lertppiriyapong, K. Mechanisms of floral repression in Arabidopsis. Curr. Opin. Plant Biol. 2003, 6, 29–35. [Google Scholar] [CrossRef]
- Amasino, R.M.; Michaels, S.D. The timing of flowering. Plant Physiol. 2010, 154, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Andrés, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Winter, C.M.; Wu, M.F.; Kanno, Y.; Yamaguchi, A.; Seo, M.; Wagner, D. Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis. Science 2014, 344, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.J. A suite of photoreceptors entrains the plant circadian clock. J. Biol. Rhythms 2003, 18, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Suárezlópez, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. Constans mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116. [Google Scholar] [CrossRef] [PubMed]
- Aukeman, M.J.; Hirschfeld, M.; Weaver, L.; Weaver, T.; Clack, T.; Amasino, R.M.; Sharrock, R.A. A deletion in the PHYD gene of the Arabidopsis Wassilewskija Ecotype defines a role for phytochrome D in red/far-red light sensing. Plant Cell 1997, 9, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Devlin, P.F.; Patel, S.R.; Whitelam, G.C. Phytochrome E influences internode elongation and flowering time in Arabidopsis. Plant Cell 1998, 10, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.L.; Beauchamp, N.; Kerckhoffs, L.H.J.; Damien Platten, J.; Reid, J.B. Interaction of phytochromes A and B in the control of de-etiolation and flower in pea. Plant J. 2001, 26, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Dean, C.; Whittaker, C. The FLC Locus: A Platform for Discoveries in Epigenetics and Adaptation. Annu. Rev. Cell Dev. Biol. 2017, 33, 555–575. [Google Scholar] [CrossRef]
- Lange, M.J.P.; Lange, T. Ovary-derived precursor gibberellin A9 is essential for female flower development in cucumber. Development 2016, 143, 4425–4429. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Z.; Zhou, Y.P.; Lv, T.X.; Xie, C.P.; Tian, C.E. Research progress on the autonomous flowering time pathway in Arabidopsis. Physiol. Mol. Biol. Plants 2017, 23, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G. The autonomous pathway: Epigenetic and posttranscriptional gene regulation in the control of Arabidopsis flowering time. Curr. Opin. Plant Biol. 2004, 7, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, S.; Boss, P.K.; Hadfield, J.; Dean, C. Additional targets of the Arabidopsis autonomous pathway members, FCA and FY. J. Exp. Bot. 2006, 57, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef] [PubMed]
- Bäurle, I.; Smith, L.; Baulcombe, D.C.; Dean, C. Widespread role for the flowering-time regulators FCA and FPA in RNA-mediated chromatin silencing. Science 2007, 318, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Bäurle, I.; Dean, C. Differential interactions of the autonomous pathway RRM proteins and chromatin regulators in the silencing of Arabidopsis targete. PLoS ONE 2008, 3, e2733. [Google Scholar] [CrossRef] [PubMed]
- Putterill, J.; Laurie, R.; Macknight, R. It’s time to flower: The genetic control of flowering time. Bioessays 2004, 26, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kaya, H.; Goto, K.; Iwabuchi, M.; Araki, T. A pair of related genes with antagonistic roles in mediating flowering signals. Science 1999, 286, 1960. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Weigel, D. Integration of floral inductive signals in Arabidopsis. Nature 2000, 404, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Chung, K.S.; Kim, J.; Lee, J.H.; Hong, S.M.; Yoo, S.J. CONSTANS Activates SUPPERSSOR OF OVEREXPRESSION OF CONSTAN 1 through FLOWERING LOCUS T to Promote Flowering in Arabidopsis. Plant Physiol. 2005, 139, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Adams, J.P.; Kim, H.J.; No, K.; Ma, C.; Strauss, S.H.; Drnevich, J.; Vandervelde, L.; Ellis, J.D.; Rice, B.M.; et al. FLOWERING LOCUS T duplication coordinates reproductive and vegetative growth in perennial poplar. Proc. Natl. Acad. Sci. USA 2011, 108, 10756–10761. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Z.; Luo, X.; Wu, C.Y.; Cao, S.Y.; Zhou, Y.F.; Jie, B.; Cao, Y.L.; Meng, H.J.; Wu, G.L. Comparative Transcriptome Analysis of Genes Involved in Anthocyanin Biosynthesis in Red and Green Walnut (Juglans regia L.). Molecules 2018, 23, 25. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Chen, W.; Fan, T.G.; Tian, Y.R.; Zhang, S.; Zeng, D.X.; Li, Y.H. Low temperature-induced DNA hypermethylation attenuates expression of RhAG, an AGAMOUS homolog, and increases petal number in rose (Rosa hybrida). BMC Plant Biol. 2015, 15, 237. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.; Wang, L.; Wu, J.; Du, H.; Liu, Z.; Ren, H.; Zhang, J. Analysis of the formation of flower shapes in wild species and cultivars of tree peony using the MADS-box subfamily gene. Gene 2012, 493, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Li, Y.L.; Zhang, Y.; Guan, S.M.; Liu, C.Y.; Zheng, G.S.; Gai, S.P. Isolation and Characterization of a SOC1-Like Gene from Tree Peony (Paeonia suffruticosa). Plant Mol. Biol. Rep. 2015, 33, 855–866. [Google Scholar] [CrossRef]
- Zhou, H.; Cheng, F.Y.; Wang, R.; Zhong, Y.; He, C.Y. Transcriptome comparison reveals key candidate genes responsible for the unusual reblooming trait in tree peonies. PLoS ONE 2013, 8, e79996. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Cheng, F.Y.; Wu, J.; He, C.Y. Isolation and functional analysis of flowering locus T in Tree Peonies (PsFT). J. Am. Soc. Hortic. Sci. 2015, 140, 265–271. [Google Scholar]
- Liu, C.L.; Zhou, Q.; Dong, L.; Wang, H.; Liu, F.; Weng, J.F.; Li, X.H.; Xie, C.X. Genetic architecture of the maize kernel row number revealed by combining QTL mapping using a high-density genetic map and bulked segregant RNA sequencing. BMC Genom. 2016, 17, 915. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, P.X.; Xu, Y.B. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, D.L.; Liu, S.Z.; Ma, X.L.; Dietrich, C.R.; Hu, H.C.; Zhang, G.S.; Liu, Z.Y.; Zheng, J.; Wang, G.Y.; et al. The maize glossy13 gene, cloned via BSR-Seq and Seq-walking encodes a putative ABC transporter required for the normal accumulation of epicuticular waxes. PLoS ONE 2013, 8, e82333. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.; Liu, S.Z.; Wen, T.J.; Paschold, A.; Marcon, C.; Tang, H.M.; Li, D.L.; Li, L.; Meeley, R.B.; Sakai, H.J.; et al. Roothairless5, which ftznctions in maize (Zea mays L.) root hair initiation and elongation encodes a monocot-specific NADPH oxidase. Plant J. 2014, 79, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Liu, S.Z.; Hill-Skinner, S.; Wu, W.; Reed, D.; Yeh, C.T.; Nettleton, D.; Schnable, P.S. The maize Brown midrib2 (bm2) gene encodes a methylenetetrahydrofolate reductase that contributes to lignin accumulation. Plant J. 2014, 77, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gonzalez, R.H.; Segovia, V.; Bird, N.; Fenwick, P.; Holdgate, S.; Berry, S.; Jack, P.; Caccamo, M.; Uauy, C. RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 2015, 13, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Livaja, M.; Wang, Y.; Wieckhorst, S.; Haseneyer, G.; Seidel, M.; Hshn, V.; Knapp, S.J.; Taudien, S.; Schön, C.C.; Bauer, E. BSTA: A targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom. 2013, 14, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Shu, Q.Y.; Wischnitzki, E.; Liu, Z.A.; Ren, H.X.; Han, X.Y.; Hao, Q.; Gao, F.F.; Xu, S.X.; Wang, L.S. Functional annotation of expressed sequence tags as a tool to understand the molecular mechanism controlling flower bud development in tree peony. Physiol. Plantarum 2009, 135, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Gai, S.P.; Zhang, Y.X.; Mu, P.; Liu, C.Y.; Liu, S.; Dong, L.; Zheng, G.S. Transcriptome analysis of tree peony during chilling requirement fulfillment: Assembling, annotation and markers discovering. Gene 2012, 497, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Y.J.; Fu, J.X.; Dong, L.; Gao, S.L.; Du, D.N. Transcriptomic analysis of cut tree peony with glucose supply using the RNA-Seq technique. Plant Cell Rep. 2014, 33, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Wang, L.S.; Shu, Q.Y.; Wu, J.; Chen, L.G.; Shao, S.; Yin, D.D. Fatty acid composition of developing tree peony (Paeonia section Moutan DC.) seeds and transcriptome analysis during seed development. BMC Genom. 2015, 16, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Cao, W.Q.; Wu, S.H.; Qian, W.F. Genetic interaction network as an important determinant of gene order in genome evolution. Mol. Biol. Evol. 2017, 34, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Guo, L.L.; Zhang, L.; Zhang, L.X.; Ma, H.L.; Guo, D.L.; Hou, X.G. Construction of a genetic linkage map in tree peony (Paeonia Sect Moutan) using simple sequence repeat (SSR) markers. Sci. Hortic. 2017, 219, 294–301. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.D.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.Z.; Lopez, R.; Martin, M.M.M.J.; et al. UniProt: The Universal Protein knowledgebase. Nucl. Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucl. Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucl. Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Sonnhammer, J.M.E.L.L.; et al. Pfam: The protein families database. Nucl. Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Colin, N.D. RSEM: Accurate transcript quantification from RNA Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.J.V.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, J.G.; Hu, Y.H.; Yang, J. Selection of Reference Genes for Quantitative Real-Time PCR during Flower Development in Tree Peony (Paeonia suffruticosa Andr.). Front. Plant Sci. 2016, 20, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Samples-ID | Base Number | Read Number | GC Content (%) | %≥Q30 | Clean Reads | Mapped Reads | Mapped Ratio |
|---|---|---|---|---|---|---|---|---|
| male parent (♂) | T-01 | 9,332,641,596 | 37,069,313 | 44.69 | 87.82% | 37,069,313 | 31,527,468 | 85.05% |
| female parent (♀) | T-02 | 8,531,057,760 | 33,883,337 | 44.82 | 87.62% | 33,883,337 | 28,497,709 | 84.11% |
| Early flowering pool | T-03 | 20,778,247,714 | 82,518,947 | 44.61 | 88.16% | 82,518,947 | 70,633,854 | 85.60% |
| Late flowering pool | T-04 | 17,887,013,878 | 71,040,683 | 44.68 | 88.02% | 71,040,683 | 60,509,445 | 85.18% |
| DEG Set | All DEG | Up-Regulated | Down-Regulated |
|---|---|---|---|
| T01- vs. -T02 | 4789 | 2309 | 2480 |
| T03- vs. -T01 | 4211 | 1983 | 2228 |
| T03- vs. -T02 | 4415 | 1932 | 2483 |
| T04- vs. -T01 | 3257 | 1398 | 1859 |
| T04- vs. -T02 | 3644 | 1484 | 2160 |
| T04- vs. -T03 | 3783 | 1879 | 1094 |
| DEG Set | Annotated | COG | GO | KEGG | KOG | Pfam | Swiss-Prot | NR |
|---|---|---|---|---|---|---|---|---|
| T01- vs. -T02 | 2606 | 815 | 1412 | 843 | 1321 | 1954 | 1816 | 2560 |
| T03- vs. -T01 | 2911 | 978 | 1663 | 920 | 1424 | 2325 | 2118 | 2872 |
| T03- vs. -T02 | 3004 | 1035 | 1754 | 960 | 1504 | 2450 | 2245 | 2972 |
| T04- vs. -T01 | 2140 | 687 | 1183 | 714 | 1070 | 1679 | 1517 | 2104 |
| T04- vs. -T02 | 2172 | 710 | 1198 | 744 | 1135 | 1703 | 1553 | 2138 |
| T04- vs. -T03 | 2993 | 1063 | 1780 | 963 | 1478 | 2507 | 2294 | 2972 |
| Unigene | Annotation | All Count | Asso Count | p-Value | FDR | Regulated | |
|---|---|---|---|---|---|---|---|
| T01- vs. -T02 | T04- vs. -T03 | ||||||
| c42942.graph_c0 | Carbohydrate transport and metabolism | 4 | 4 | 0 | 0 | down | down |
| c46352.graph_c0 | Plant invertase/pectin methylesterase inhibitor | 4 | 4 | 0 | 0 | up | down |
| c58332.graph_c0 | K+ potassium transporter | 9 | 7 | 0 | 0 | up | down |
| c58361.graph_c0 | Hothead-like | 13 | 12 | 0 | 0 | down | down |
| c57417.graph_c0 | Reduced wall acetylation 4-like | 16 | 5 | 1.23 × 10−9 | 1.41 × 10−6 | down | down |
| c58526.graph_c0 | PTI 1-like tyrosine-protein kinase | 14 | 4 | 4.19 × 10−8 | 2.10 × 10−5 | down | up |
| c53143.graph_c0 | peptidyl-prolyl cis-trans isomerase activity | 8 | 3 | 2.05 × 10−7 | 9.16 × 10−5 | up | up |
| Searching Item | Number |
|---|---|
| Total number of sequences examined | 16,885 |
| Total size of examined sequences (bp) | 33,407,765 |
| Total number of identified SSRs | 6678 |
| Number of SSR containing sequences | 5191 |
| Number of sequences containing more than 1 SSR | 1170 |
| Number of SSRs present in compound formation | 333 |
| Mono nucleotide | 4347 |
| Di nucleotide | 1443 |
| Tri nucleotid | 833 |
| Tetra nucleotide | 35 |
| Penta nucleotide | 8 |
| Hexa nucleotide | 12 |
| Samples | Homozygosity | Heterozygosity | SNP Number |
|---|---|---|---|
| T01 | 109,252 | 81,140 | 190,392 |
| T02 | 142,760 | 43,169 | 185,929 |
| T03 | 40,888 | 166,550 | 207,438 |
| T04 | 50,735 | 155,675 | 206,410 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, X.; Guo, Q.; Wei, W.; Guo, L.; Guo, D.; Zhang, L. Screening of Genes Related to Early and Late Flowering in Tree Peony Based on Bulked Segregant RNA Sequencing and Verification by Quantitative Real-Time PCR. Molecules 2018, 23, 689. https://doi.org/10.3390/molecules23030689
Hou X, Guo Q, Wei W, Guo L, Guo D, Zhang L. Screening of Genes Related to Early and Late Flowering in Tree Peony Based on Bulked Segregant RNA Sequencing and Verification by Quantitative Real-Time PCR. Molecules. 2018; 23(3):689. https://doi.org/10.3390/molecules23030689
Chicago/Turabian StyleHou, Xiaogai, Qi Guo, Weiqiang Wei, Lili Guo, Dalong Guo, and Lin Zhang. 2018. "Screening of Genes Related to Early and Late Flowering in Tree Peony Based on Bulked Segregant RNA Sequencing and Verification by Quantitative Real-Time PCR" Molecules 23, no. 3: 689. https://doi.org/10.3390/molecules23030689
APA StyleHou, X., Guo, Q., Wei, W., Guo, L., Guo, D., & Zhang, L. (2018). Screening of Genes Related to Early and Late Flowering in Tree Peony Based on Bulked Segregant RNA Sequencing and Verification by Quantitative Real-Time PCR. Molecules, 23(3), 689. https://doi.org/10.3390/molecules23030689