
Performance of Different Immobilized Lipases in the Syntheses of Short- and Long-Chain Carboxylic Acid Esters by Esterification Reactions in Organic Media

 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
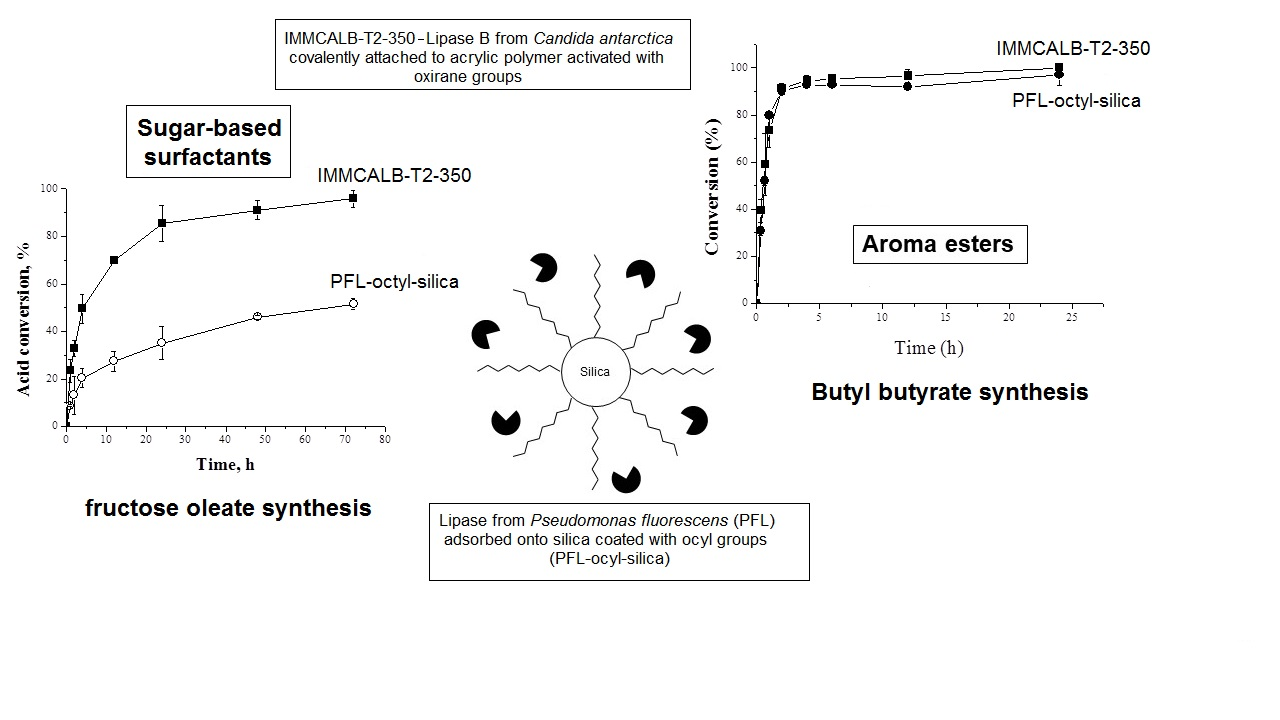
2.1. Synthesis of Aroma Esters
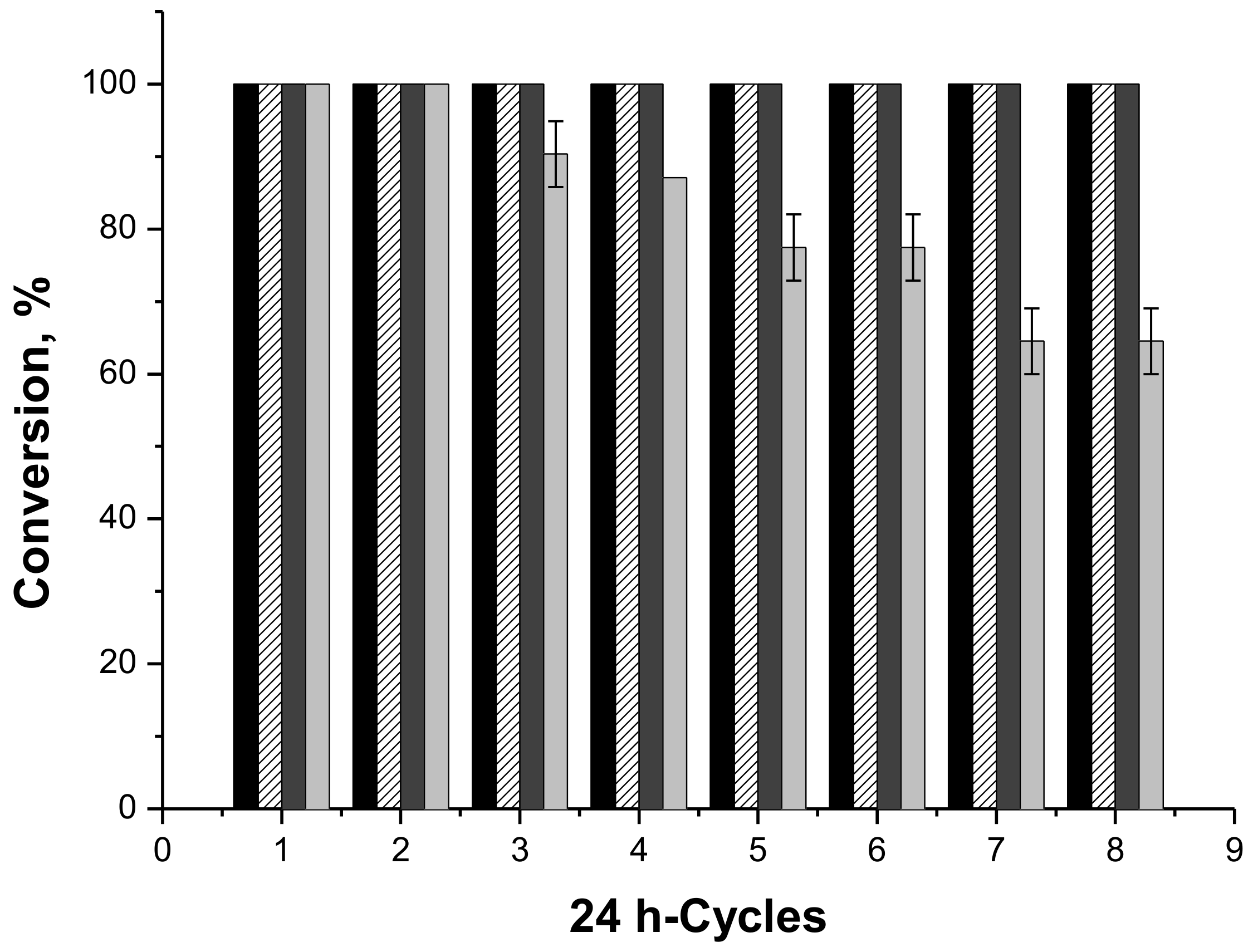
2.2. Operational Stability of the Different Biocatalyst in the Synthesis of Butyl Butyrate
2.3. Reaction Course of Butyl Butyrate Synthesis by PFL-Octyl-Silica and IMMCALB-T2-350 Biocatalysts
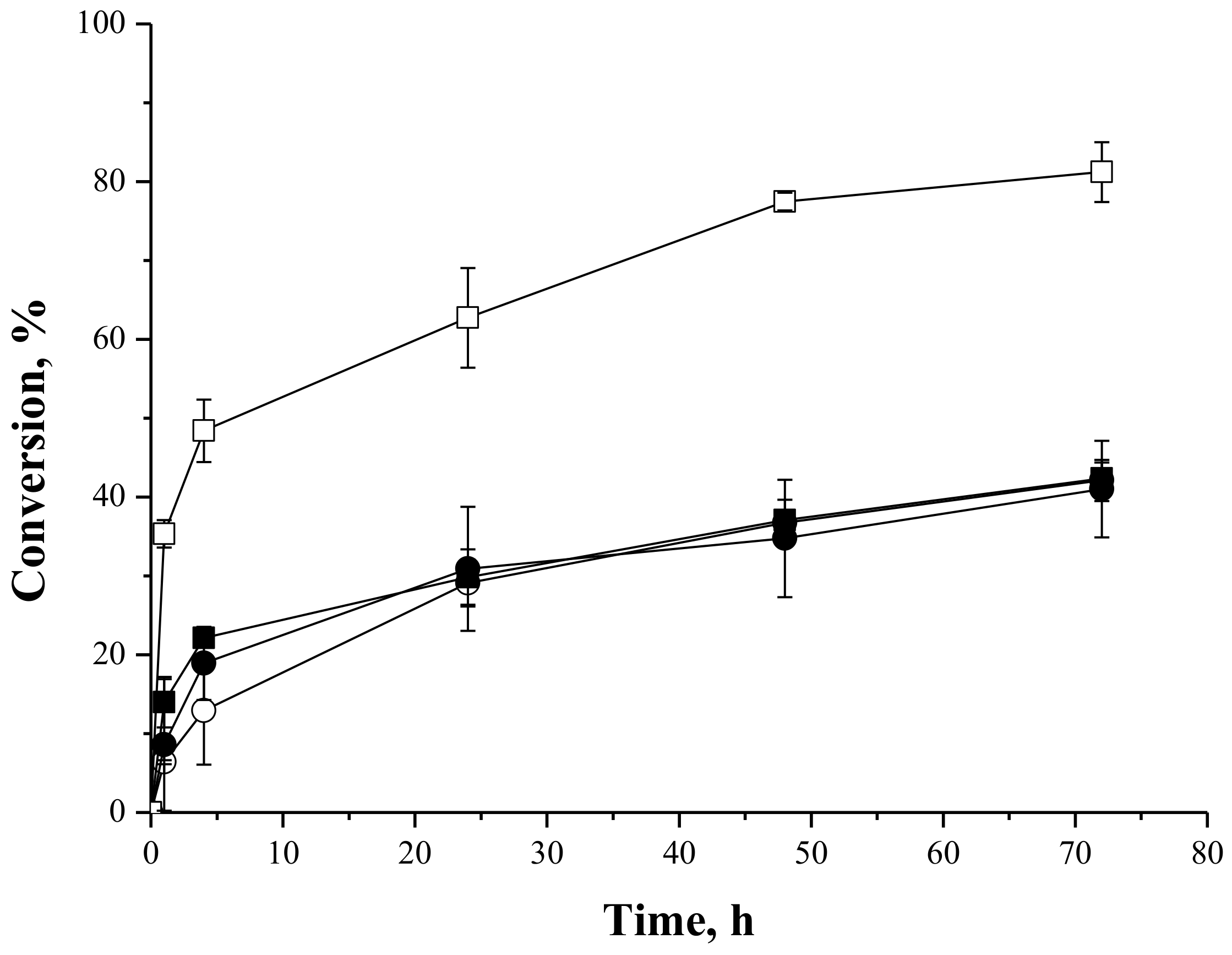
2.4. Syntheses of Sugar Esters
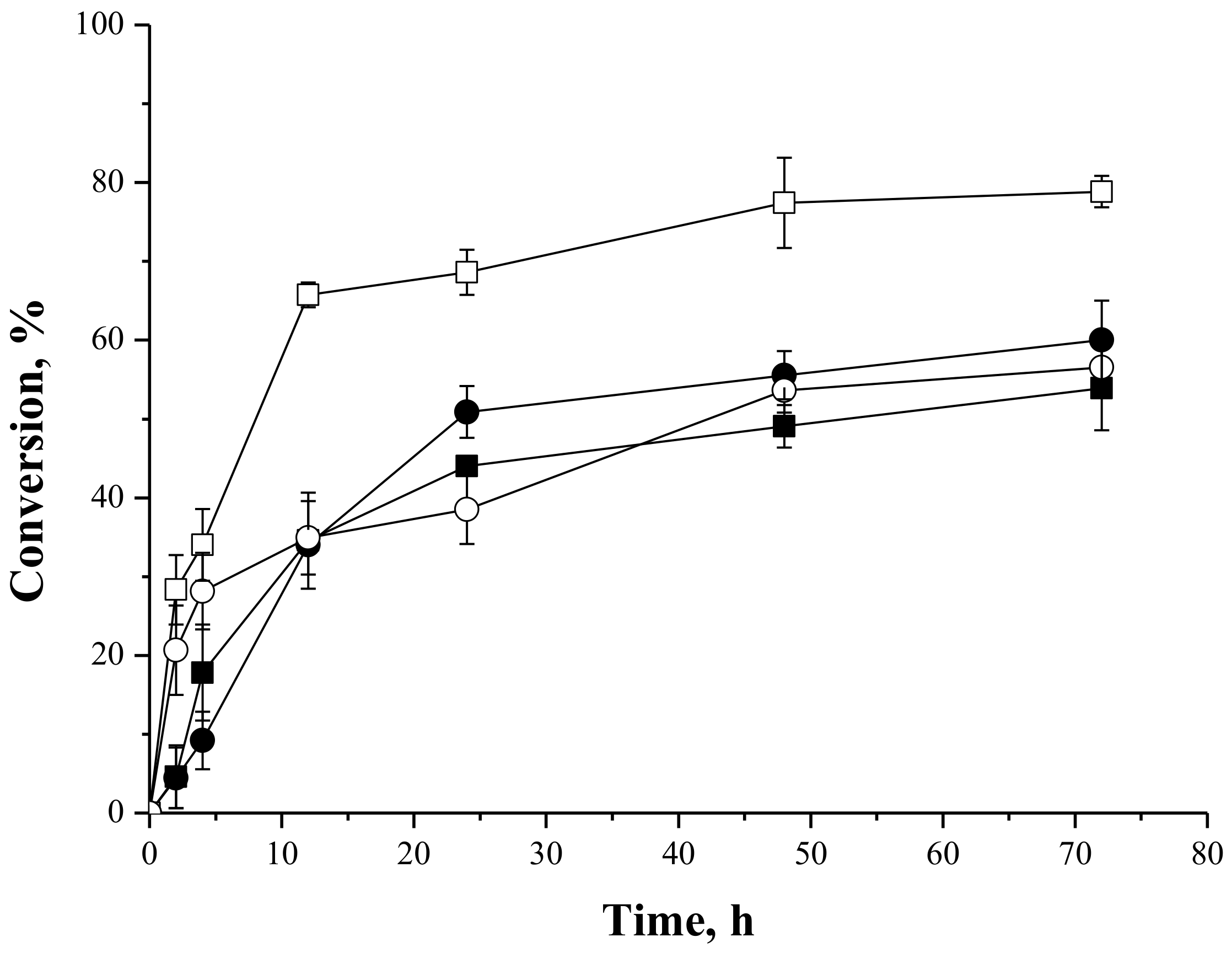
2.4.1. Syntheses of Fructose Oleate—Stoichiometric Ratio
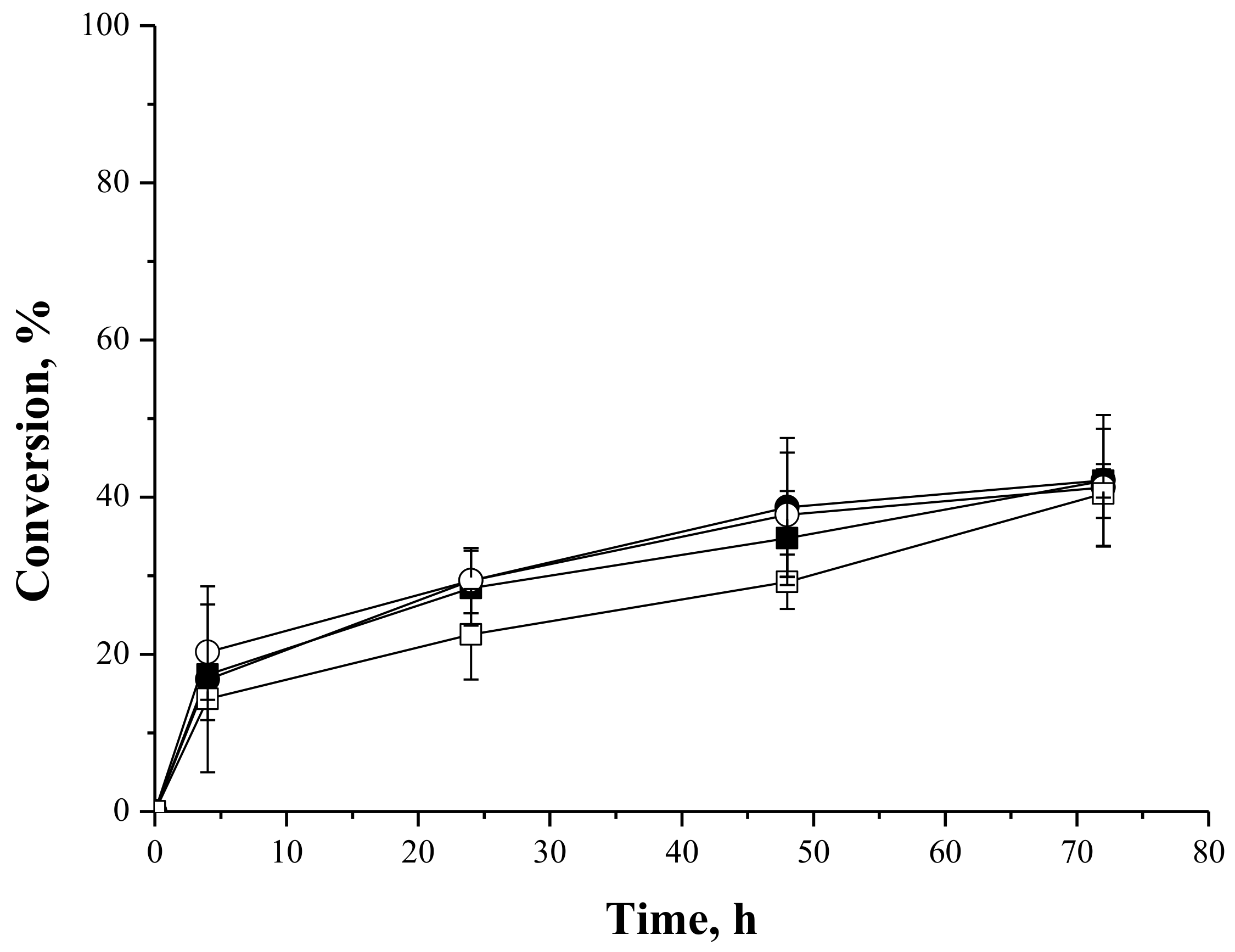
2.4.2. Syntheses of Lactose Oleate—Stoichiometric Ratio
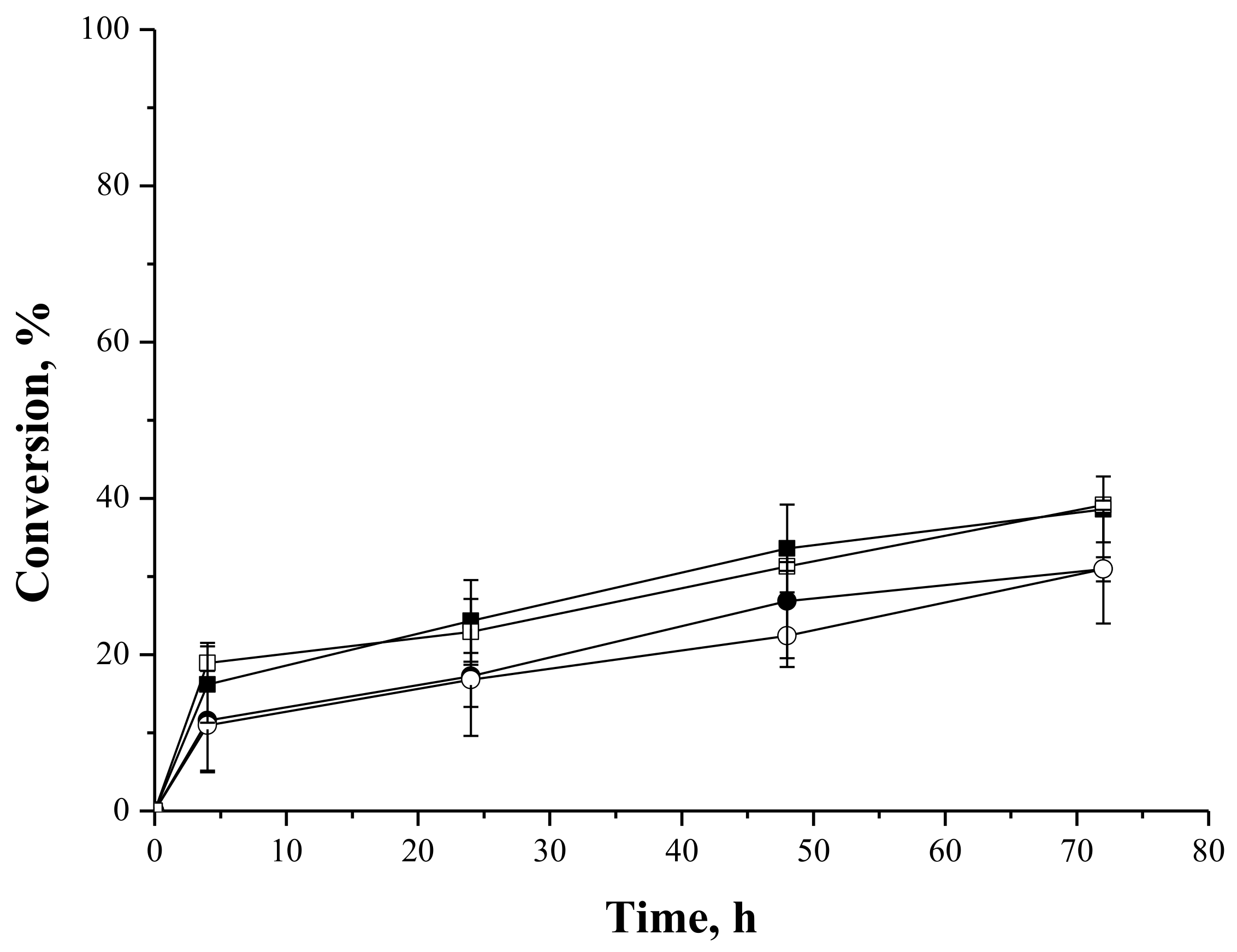
2.4.3. Syntheses of Fructose and Lactose Laurate—Stoichiometric Ratio
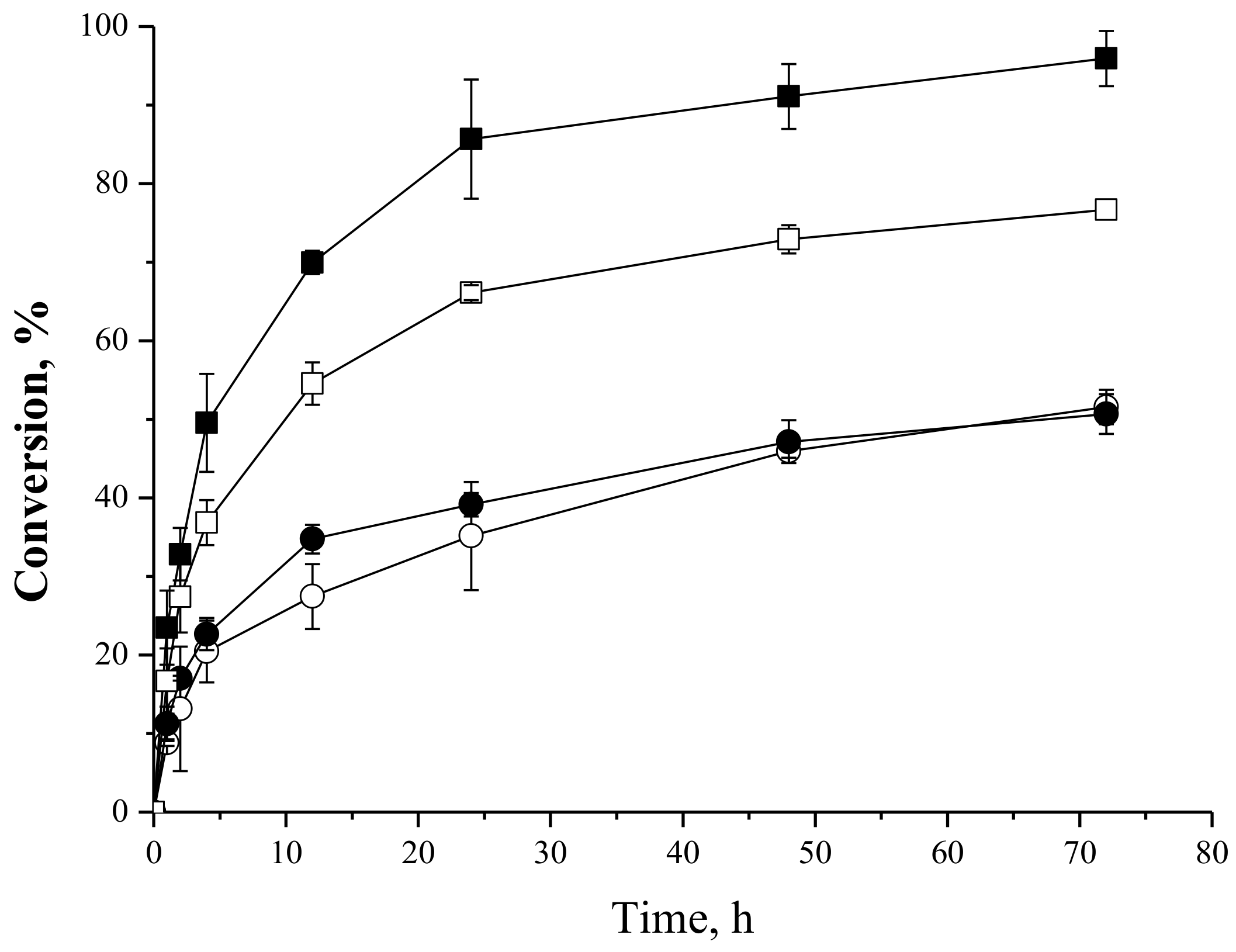
2.4.4. Effect of the Fatty Acid:Carbohydrate Molar Ratio in the Synthesis of Oleate Fructose Esters
2.4.5. Effect of the Biocatalyst Mass
2.4.6. Operational Stability of the Biocatalysts in the Syntheses of Fructose Oleate
3. Materials and Methods
3.1. Materials
3.2. Production of Octyl-Silica Support
3.3. Immobilization of PFL on Octyl-Silica
3.4. Synthesis of Flavor Esters
3.5. Synthesis of Sugar Esters
3.6. Operational Stability of the Biocatalysts
3.6.1. Flavor Ester (Butyl Butyrate)
3.6.2. Sugar Ester (Fructose Oleate)
3.7. Esterification Activity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, S.-Q.; Holland, R.; Crow, V.L. Esters and their biosynthesis in fermented dairy products: A review. Int. Dairy J. 2004, 14, 923–945. [Google Scholar] [CrossRef]
- Sumby, K.M.; Grbin, P.R.; Jiranek, V. Microbial modulation of aromatic esters in wine: Current knowledge and future prospects. Food Chem. 2010, 121, 1–16. [Google Scholar] [CrossRef]
- Abbas, H.; Comeau, L. Aroma synthesis by immobilized lipase from Mucor sp. Enzym. Microb. Technol. 2003, 32, 589–595. [Google Scholar] [CrossRef]
- Khan, N.R.; Rathod, V.K. Enzyme catalyzed synthesis of cosmetic esters and its intensification: A review. Process Biochem. 2015, 50, 1793–1806. [Google Scholar] [CrossRef]
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzym. Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Kiss, M.A.; Sefanovits-Bányai, É.; Tóth, Á.; Boross, L. Extractive Synthesis of Ethyl-Oleate Using Alginate Gel Co-Entrapped Yeast Cells and Lipase Enzyme. Eng. Life Sci. 2004, 4, 460–464. [Google Scholar] [CrossRef]
- Kobayashi, T. Lipase-catalyzed syntheses of sugar esters in non-aqueous media. Biotechnol. Lett. 2011, 33, 1911–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumel, A.M.; Annuar, M.S.M.; Heidelberg, T.; Chisti, Y. Lipase mediated synthesis of sugar fatty acid esters. Process Biochem. 2011, 46, 2079–2090. [Google Scholar] [CrossRef]
- Kosaric, N. Biosurfactants in industry. Pure Appl. Chem. 1992, 64, 1731–1737. [Google Scholar] [CrossRef]
- Bidjou-Haiour, C.; Klai, N. Lipase catalyzed synthesis of fatty acid xylose esters and their surfactant properties. Asian J. Chem. 2013, 25, 4347–4350. [Google Scholar] [CrossRef]
- Patil, D.; De Leonardis, A.; Nag, A. Synthesis of Biosurfactants From Natural Resources. J. Food Biochem. 2011, 35, 747–758. [Google Scholar] [CrossRef]
- Sutili, F.K.; Ruela, H.S.; Leite, S.G.F.; Miranda, L.S.D.M.; Leal, I.C.R.; de Souza, R.O.M.A. Lipase-catalyzed esterification of steric hindered fructose derivative by continuous flow and batch conditions. J. Mol. Catal. B Enzym. 2013, 85, 37–42. [Google Scholar] [CrossRef]
- Zhang, X.; Nie, K.; Wang, M.; Liu, L.; Li, K.; Wang, F.; Tan, T.; Deng, L. Site-specific xylitol dicaprate ester synthesized by lipase from Candida sp. 99–125 with solvent-free system. J. Mol. Catal. B Enzym. 2013, 89, 61–66. [Google Scholar] [CrossRef]
- Ferrer, M.; Soliveri, J.; Plou, F.J.; López-Cortés, N.; Reyes-Duarte, D.; Christensen, M.; Copa-Patiño, J.L.; Ballesteros, A. Synthesis of sugar esters in solvent mixtures by lipases from Thermomyces lanuginosus and Candida antarctica B, and their antimicrobial properties. Enzym. Microb. Technol. 2005, 36, 391–398. [Google Scholar] [CrossRef]
- Yan, Y.; Bornscheuer, U.T.; Stadler, G.; Lutz-Wahl, S.; Otto, R.T.; Reuss, M.; Schmid, R.D. Regioselective lipase-catalyzed synthesis of glucose ester on a preparative scale. Eur. J. Lipid Sci. Technol. 2001, 103, 583–587. [Google Scholar] [CrossRef]
- Boscolo, M. Sucrochemistry: Synthesis and potentialities for applications of some sucrose chemical derivates. Quim. Nova 2003, 26, 906–912. [Google Scholar] [CrossRef]
- El Seoud, O.A.; Koschella, A.; Fidale, L.C.; Dorn, S.; Heinze, T. Applications of ionic liquids in carbohydrate chemistry: A window of opportunities. Biomacromolecules 2007, 8, 2629–2647. [Google Scholar] [CrossRef] [PubMed]
- Abdulmalek, E.; Hamidon, N.F.; Abdul Rahman, M.B. Optimization and characterization of lipase catalysed synthesis of xylose caproate ester in organic solvents. J. Mol. Catal. B Enzym. 2016, 132, 1–4. [Google Scholar] [CrossRef]
- De Castro, H.F.; Mendes, A.A.; Dos Santos, J.C.; De Aguiar, C.L. Modificação de óleos e gorduras por biotransformação. Quim. Nova 2004, 27, 146–156. [Google Scholar] [CrossRef]
- Kumari, A.; Mahapatra, P.; Garlapati, V.K.; Banerjee, R. Enzymatic transesterification of Jatropha oil. Biotechnol. Biofuels 2009, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Fallavena, L.P.; Antunes, F.H.F.; Alves, J.S.; Paludo, N.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Ultrasound technology and molecular sieves improve the thermodynamically controlled esterification of butyric acid mediated by immobilized lipase from Rhizomucor miehei. RSC Adv. 2014, 4, 8675. [Google Scholar] [CrossRef]
- Mai, N.L.; Ahn, K.; Bae, S.W.; Shin, D.W.; Morya, V.K.; Koo, Y.-M. Ionic liquids as novel solvents for the synthesis of sugar fatty acid ester. Biotechnol. J. 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.G.; Li, J.R.; Chu, Y.H. Enzyme-catalyzed regioselective synthesis of sucrose-based esters. J. Chem. Technol. Biotechnol. 2011, 86, 1457–1468. [Google Scholar] [CrossRef]
- Reetz, M.T. Lipases as practical biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 145–150. [Google Scholar] [CrossRef]
- Abdulmalek, E.; Mohd Saupi, H.S.; Tejo, B.A.; Basri, M.; Salleh, A.B.; Raja Abd Rahman, R.N.Z.; Abdul Rahman, M.B. Improved enzymatic galactose oleate ester synthesis in ionic liquids. J. Mol. Catal. B Enzym. 2012, 76, 37–43. [Google Scholar] [CrossRef]
- Vescovi, V.; dos Santos, J.B.C.; Tardioli, P.W. Porcine pancreatic lipase hydrophobically adsorbed on octyl-silica: A robust biocatalyst for syntheses of xylose fatty acid esters. Biocatal. Biotransform. 2017, 35. [Google Scholar] [CrossRef]
- Vescovi, V.; Kopp, W.; Guisán, J.M.; Giordano, R.L.C.; Mendes, A.A.; Tardioli, P.W. Improved catalytic properties of Candida antarctica lipase B multi-attached on tailor-made hydrophobic silica containing octyl and multifunctional amino- glutaraldehyde spacer arms. Process Biochem. 2016, 51, 2055–2066. [Google Scholar] [CrossRef]
- Vescovi, V.; Giordano, R.L.C.; Mendes, A.A.; Tardioli, P.W. Immobilized Lipases on Functionalized Silica Particles as Potential Biocatalysts for the Synthesis of Fructose Oleate in an Organic Solvent/Water System. Molecules 2017, 22, 212. [Google Scholar] [CrossRef] [PubMed]
- Chamouleau, F.; Coulon, D.; Girardin, M.; Ghoul, M. Influence of water activity and water content on sugar esters lipase-catalyzed synthesis in organic media. J. Mol. Catal. B Enzym. 2001, 11, 949–954. [Google Scholar] [CrossRef]
- Chang, S.W.; Shaw, J.F. Biocatalysis for the production of carbohydrate esters. New Biotechnol. 2009, 26, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Coulon, D.; Ismail, A.; Girardin, M.; Rovel, B.; Ghoul, M. Effect of different biochemical parameters on the enzymatic synthesis of fructose oleate. J. Biotechnol. 1996, 51, 115–121. [Google Scholar] [CrossRef]
- Degn, P.; Zimmermann, W. Optimization of carbohydrate fatty acid ester synthesis in organic media by a lipase from Candida antarctica. Biotechnol. Bioeng. 2001, 74, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Neta, N.S.; Teixeira, J.A.; Rodrigues, L.R. Sugar Ester Surfactants: Enzymatic Synthesis and Applications in Food Industry. Crit. Rev. Food Sci. Nutr. 2015, 55, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Šabeder, S.; Habulin, M.; Knez, Ž. Lipase-catalyzed synthesis of fatty acid fructose esters. J. Food Eng. 2006, 77, 880–886. [Google Scholar] [CrossRef]
- Tarahomjoo, S.; Alemzadeh, I. Surfactant production by an enzymatic method. Enzym. Microb. Technol. 2003, 33, 33–37. [Google Scholar] [CrossRef]
- Bezbradica, D.; Mijin, D.; Šiler-Marinković, S.; Knežević, Z. The effect of substrate polarity on the lipase-catalyzed synthesis of aroma esters in solvent-free systems. J. Mol. Catal. B Enzym. 2007, 45, 97–101. [Google Scholar] [CrossRef]
- Verger, R. ‘’Interfacial activation’’ of lipases : Facts and artifacts. Trends Biotechnol. 1997, 15, 32–38. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Derewenda, U.; Derewenda, Z.S.; Dodson, G.G.; Lawson, D.M.; Turkenburg, J.P.; Bjorkling, F.; Huge-Jensen, B.; Patkar, S.A.; Thim, L. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature 1991, 351, 491. [Google Scholar] [CrossRef] [PubMed]
- Bastida, A.; Sabuquillo, P.; Armisen, P.; Fernández-Lafuente, R.; Huguet, J.; Guisán, J.M. A single step purification, immobilization, and hyperactivation of lipases via interfacial adsorption on strongly hydrophobic supports. Biotechnol. Bioeng. 1998, 58, 486–493. [Google Scholar] [CrossRef]
- Palomo, J.M.; Muñoz, G.; Fernández-Lorente, G.; Mateo, C.; Fernández-Lafuente, R.; Guisán, J.M. Interfacial adsorption of lipases on very hydrophobic support (octadecyl–Sepabeads): Immobilization, hyperactivation and stabilization of the open form of lipases. J. Mol. Catal. B Enzym. 2002, 19–, 279–286. [Google Scholar] [CrossRef]
- Martinelle, M.; Holmquist, M.; Hult, K. On the interfacial activation of Candida antarctica lipase A and B as compared with Humicola lanuginosa lipase. Biochim. Biophys. Acta 1995, 1258, 272–276. [Google Scholar] [CrossRef]
- Palomo, J.M.; Ortiz, C.; Fuentes, M.; Fernández-Lorente, G.; Gusian, J.M.; Fernandez-Lafuente, R. Use of immobilized lipases for lipase purification via specific lipase–lipase interactions. J. Chromatogr. A 2004, 1038, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M.; Ortiz, C.; Fernández-Lorente, G.; Fuentes, M.; Guisán, J.M.; Fernández-Lafuente, R. Lipase–lipase interactions as a new tool to immobilize and modulate the lipase properties. Enzym. Microb. Technol. 2005, 36, 447–454. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R.; Armisén, P.; Sabuquillo, P.; Fernández-Lorente, G.; Guisán, J.M. Immobilization of lipases by selective adsorption on hydrophobic supports. Chem. Phys. Lipids 1998, 93, 185–197. [Google Scholar] [CrossRef]
- Lima, L.N.; Aragon, C.C.; Mateo, C.; Palomo, J.M.; Giordano, R.L.C.; Tardioli, P.W.; Guisan, J.M.; Fernandez-Lorente, G. Immobilization and stabilization of a bimolecular aggregate of the lipase from Pseudomonas fluorescens by multipoint covalent attachment. Process Biochem. 2013, 48, 118–123. [Google Scholar] [CrossRef]
- Manoel, E.A.; dos Santos, J.C.S.; Freire, D.M.G.; Rueda, N.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports involves the open form of the enzyme. Enzym. Microb. Technol. 2015, 71. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Kobayashi, T. Synthesis of esters by immobilized-lipase-catalyzed condensation reaction of sugars and fatty acids in water-miscible organic solvent. J. Biosci. Bioeng. 2005, 99, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.L.; Balcão, V.M.; Malcata, F.X. Kinetics and mechanisms of reactions catalyzed by immobilized lipases. Enzym. Microb. Technol. 2000, 27, 187–204. [Google Scholar] [CrossRef]
- Alves, J.S.; Garcia-Galan, C.; Schein, M.F.; Silva, A.M.; Barbosa, O.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Combined effects of ultrasound and immobilization protocol on butyl acetate synthesis catalyzed by CALB. Molecules 2014, 19, 9562–9576. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.B.; Schein, M.F.; Friedrich, J.L.R.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Ultrasound-assisted butyl acetate synthesis catalyzed by Novozym 435: Enhanced activity and operational stability. Ultrason. Sonochem. 2013, 20, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Graebin, N.G.; Martins, A.B.; Lorenzoni, A.S.G.; Garcia-Galan, C.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Immobilization of lipase B from Candida antarctica on porous styrene-divinylbenzene beads improves butyl acetate synthesis. Biotechnol. Prog. 2012, 28, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Paludo, N.; Alves, J.S.; Altmann, C.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. The combined use of ultrasound and molecular sieves improves the synthesis of ethyl butyrate catalyzed by immobilized Thermomyces lanuginosus lipase. Ultrason. Sonochem. 2015, 22, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Zongliang, D.; Yao, Y.; Ruixia, L.; Dacheng, W. Effect of molecular sieves on lipase-catalyzed esterification of rutin with stearic acid. J. Agric. Food Chem. 2006, 54, 6219–6225. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, J.; Giacometti, F.; Milin, Č.; Vasić-Rački, D. Kinetic characterisation of enzymatic esterification in a solvent system: Adsorptive control of water with molecular sieves. J. Mol. Catal. B Enzym. 2001, 11, 921–928. [Google Scholar] [CrossRef]
- Friedrich, J.L.R.; Pena, F.P.; Garcia-Galan, C.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Effect of immobilization protocol on optimal conditions of ethyl butyrate synthesis catalyzed by lipase B from Candida antarctica. J. Chem. Technol. Biotechnol. 2013, 88, 1089–1095. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Palomo, J.M.; Cocca, J.; Mateo, C.; Moro, P.; Terreni, M.; Fernandez-Lafuente, R.; Guisan, J.M. Regio-selective deprotection of peracetylated sugars via lipase hydrolysis. Tetrahedron 2003, 59, 5705–5711. [Google Scholar] [CrossRef]
- Fernandez-Lafuente, R. Lipase from Thermomyces lanuginosus: Uses and prospects as an industrial biocatalyst. J. Mol. Catal. B Enzym. 2010, 62, 197–212. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Illanes, A.; Wilson, L. Heterofunctional hydrophilic-hydrophobic porous silica as support for multipoint covalent immobilization of lipases: Application to lactulose palmitate synthesis. Langmuir 2014, 30, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.N.; Oliveira, G.C.; Rojas, M.J.; Castro, H.F.; Da Rós, P.C.M.; Mendes, A.A.; Giordano, R.L.C.; Tardioli, P.W. Immobilization of Pseudomonas fluorescens lipase on hydrophobic supports and application in biodiesel synthesis by transesterification of vegetable oils in solvent-free systems. J. Ind. Microbiol. Biotechnol. 2015, 42, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.M.; Terreros, P.; Fernández-Pérez, M.; Otero, C.; Díaz-González, G. Functionalization of mesoporous silica for lipase immobilization: Characterization of the support and the catalysts. J. Mol. Catal. B Enzym. 2004, 30, 83–93. [Google Scholar] [CrossRef]
- Séverac, E.; Galy, O.; Turon, F.; Pantel, C.A.; Condoret, J.-S.; Monsan, P.; Marty, A. Selection of CalB immobilization method to be used in continuous oil transesterification: Analysis of the economical impact. Enzym. Microb. Technol. 2011, 48, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Do Neta, N.A.S.; dos Santos, J.C.S.; de Sancho, S.O.; Rodrigues, S.; Gonçalves, L.R.B.; Rodrigues, L.R.; Teixeira, J.A. Enzymatic synthesis of sugar esters and their potential as surface-active stabilizers of coconut milk emulsions. Food Hydrocoll. 2012, 27, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.K.; Bombyk, R.A.; Wagh, A.; Bingham, A.; Berreau, L.M. Synthesis of lactose monolaurate as influenced by various lipases and solvents. J. Mol. Catal. B Enzym. 2009, 60, 171–177. [Google Scholar] [CrossRef]
- Zaidan, U.H.; Abdul Rahman, M.B.; Othman, S.S.; Basri, M.; Abdulmalek, E.; Abdul Rahman, R.N.Z.R.; Salleh, A.B. Biocatalytic production of lactose ester catalysed by mica-based immobilised lipase. Food Chem. 2012, 131, 199–205. [Google Scholar] [CrossRef]
- Rueda, N.; Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved performance of lipases immobilized on heterofunctional octyl-glyoxyl agarose beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Fernandez-Lafuente, R. Evaluation of different immobilized lipases in transesterification reactions using tributyrin: Advantages of the heterofunctional octyl agarose beads. J. Mol. Catal. B Enzym. 2016, 133, 117–123. [Google Scholar] [CrossRef]
- Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Hirata, D.B.; Torrestiana-Sanchez, B.; Rosales-Quintero, A.; Fernandez-Lafuente, R. Relevance of substrates and products on the desorption of lipases physically adsorbed on hydrophobic supports. Enzym. Microb. Technol. 2017, 96, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Sánchez-Montero, J.M.; Garcia-Verdugo, E.; Porcar, R.; Fernandez-Lafuente, R. Advantages of Heterofunctional Octyl Supports: Production of 1,2-Dibutyrin by Specific and Selective Hydrolysis of Tributyrin Catalyzed by Immobilized Lipases. ChemistrySelect 2016, 1, 3259–3270. [Google Scholar] [CrossRef]
- Tani, K.; Suzuki, Y. Influence of titania matrix on retention behaviour in reversed-phase liquid chromatography. J. Chromatogr. A 1996, 722, 129–134. [Google Scholar] [CrossRef]
- De Paula, A.V.; de Souza Barboza, J.C.; de Castro, H.F. Estudo da influência do solvente, carboidrato e ácido graxo na síntese enzimática de ésteres de açúcares. Quim. Nova 2005, 28, 792–796. [Google Scholar] [CrossRef]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactants and Products | Conversions (%) by Biocatalyst | ||||||
|---|---|---|---|---|---|---|---|
| Acyl donor | Acyl acceptor | Ester | FEMA flavor profile 1 | PFL-octyl-silica | IMMTLL-T2-150 | IMMAPF-T2-150 | IMMCALB-T2-350 |
| Acetic acid | 1-butanol | Butyl acetate | apple, banana, pungent | 96.8 ± 0.1 | 97.0 ± 0.0 | 93.8 ± 0.4 | 96.8 ± 0.1 |
| Acetic acid | 1-hexanol | Hexyl acetate | apple, banana, grass, herb, pear | 98.0 ± 2.9 | 80.3 ± 1.5 | 96.9 ± 0.1 | 97.5 ± 3.5 |
| Acetic acid | isoamyl alcohol | Isoamyl acetate | Apple, banana, glue, pear | 96.9 ± 0.2 | 91.5 ± 0.0 | 93.7 ± 0.2 | 95.0 ± 2.4 |
| Butyric acid | 1-butanol | Butyl butyrate | floral | 98.4 ± 2.3 | 93.5 ± 0.0 | 96.9 ± 0.1 | 96.6 ± 0.1 |
| Acetic acid | ethanol | Ethyl acetate | Aromatic, brandy, grape | 96.7 ± 0.1 | 90.8 ± 0.2 | 88.9 ± 1.9 | 90.5 ± 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lima, L.N.; Mendes, A.A.; Fernandez-Lafuente, R.; Tardioli, P.W.; Giordano, R.D.L.C. Performance of Different Immobilized Lipases in the Syntheses of Short- and Long-Chain Carboxylic Acid Esters by Esterification Reactions in Organic Media. Molecules 2018, 23, 766. https://doi.org/10.3390/molecules23040766
De Lima LN, Mendes AA, Fernandez-Lafuente R, Tardioli PW, Giordano RDLC. Performance of Different Immobilized Lipases in the Syntheses of Short- and Long-Chain Carboxylic Acid Esters by Esterification Reactions in Organic Media. Molecules. 2018; 23(4):766. https://doi.org/10.3390/molecules23040766
Chicago/Turabian StyleDe Lima, Lionete Nunes, Adriano Aguiar Mendes, Roberto Fernandez-Lafuente, Paulo Waldir Tardioli, and Raquel De Lima Camargo Giordano. 2018. "Performance of Different Immobilized Lipases in the Syntheses of Short- and Long-Chain Carboxylic Acid Esters by Esterification Reactions in Organic Media" Molecules 23, no. 4: 766. https://doi.org/10.3390/molecules23040766
APA StyleDe Lima, L. N., Mendes, A. A., Fernandez-Lafuente, R., Tardioli, P. W., & Giordano, R. D. L. C. (2018). Performance of Different Immobilized Lipases in the Syntheses of Short- and Long-Chain Carboxylic Acid Esters by Esterification Reactions in Organic Media. Molecules, 23(4), 766. https://doi.org/10.3390/molecules23040766