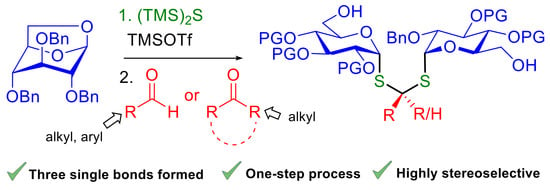
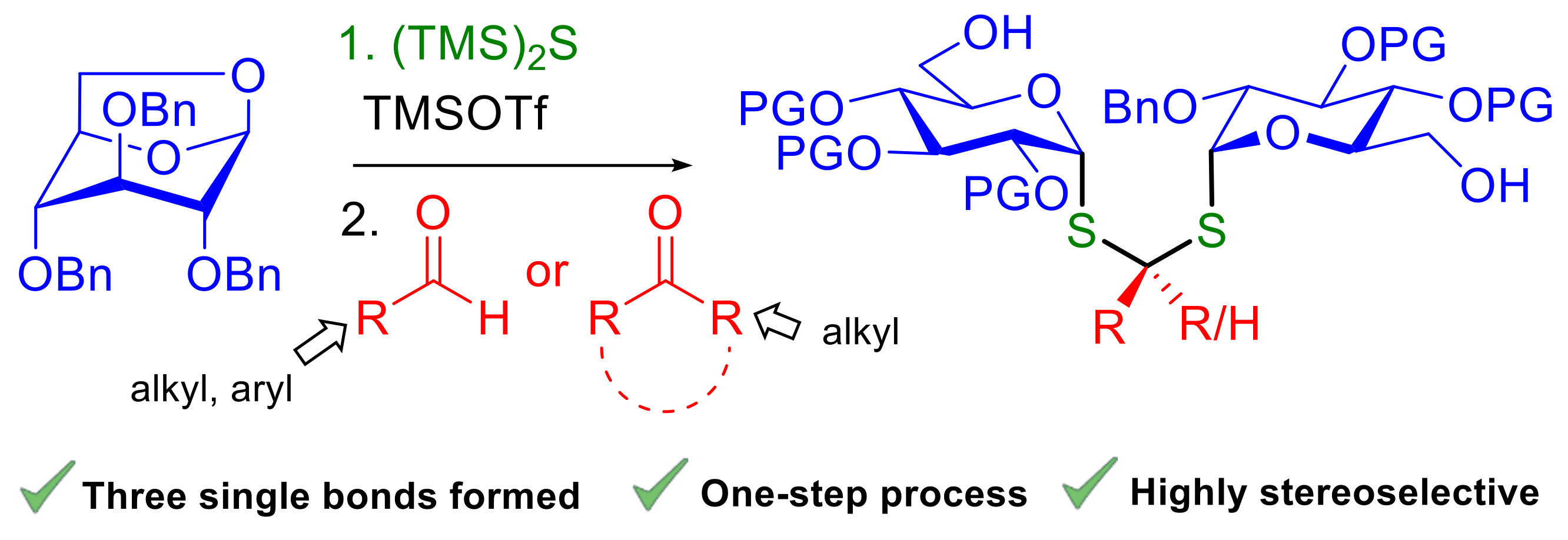
3.2. General Procedure for the Synthesis of Diglycoside Thioacetals 6
Molecular sieves 4Å (80 mg) were added to a tube. A solution of anhydroglucose 5 (1 equiv., 0.233 mmol) in CH2Cl2 (1 mL) was added via cannula to the flask. Bis (trimethylsilyl) sulfide (1.4 equiv., 58.3 mg, 0.0613 mL, 0.327 mmol) followed by trimethylsilyl trifluoromethane sulfonate (1.1 equiv., 57 mg, 0.0466 mL, 0.257 mmol) were added to the mixture. The tube was sealed and the medium was stirred at 60 °C. After 2 h of reaction, a solution of the corresponding aldehyde (1 equiv., 0.233 mmol) in 0.3 mL of CH2Cl2 was added at −30 °C. The reaction was stirred for 1 h 30 min. The mixture was warmed up to r.t. and washed with saturated aqueous NaHCO3 (50 mL). The aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The organic layers were combined and washed with brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure. The crude obtained was purified by flash column chromatography (Petroleum Ether/EtOAc) to afford compounds 6.
Dithioacetal 6a. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6a was obtained as a solid in 60% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 4/6). + 177 (c 0.7, CHCl3). IR 3431 cm-1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.44–7.22 (m, 35H, Ph), 5.95 (d, J = 5.5 Hz, 1H, H-1 or H-1′), 5.02 (s, 1H, H-7), 5.00–4.97 (m, 2H, O-CH2-Ph, H-1 or H-1′), 4.91–4.77 (m, 5H, O-CH2-Ph), 4.71 (d, J = 10.8 Hz, 1H, O-CH2-Ph), 4.62 (d, J = 11.4 Hz, 1H, O-CH2-Ph), 4.57 (d, J = 5.9 Hz, 1H, O-CH2-Ph), 4.54 (d, J = 5.7 Hz, 1H, O-CH2-Ph), 4.38 (d, J = 11.5 Hz, 1H, O-CH2-Ph), 4.32 (d, J = 11.5 Hz, 1H, O-CH2-Ph), 4.21 (m, 1H, H-5 or H-5′), 4.14 (m, 1H, H-5 or H-5′), 3.98–3.80 (m, 5H, H-2 or H-2′, H-6, H-6′, H3, H3′), 3.67 (dd, J = 9.5; 5.6 Hz, 1H, H-2 or H-2′), 3.63–3.55 (m, 2H, H-6, H6′), 3.37 (dd, J = 10.0; 8.8 Hz, 1H, H-4 or H-4′), 3.28 (dd, J = 9.9; 8.8 Hz, 1H, H-4 or H-4′). 13C NMR (100 MHz, CDCl3) δ (ppm) 139.4 (Cq-Ar), 138.6 (Cq-Ar), 138.55 (Cq-Ar), 138.1 (Cq-Ar), 137.8 (Cq-Ar), 137.7 (Cq-Ar), 137.3 (Cq-Ar), 128.9–127.8 (35 × CHAr) 82.8 (CH) 82.7 (CH), 82.6 (CH), 81.9 (C-1 or C-1′), 79.4 (CH), 78.8 (CH), 77.8 (C-4 or C-4′), 78.4 (C-4 or C-4′), 75.9 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.2 (O-CH2-Ph), 72.8 (C5 or C-5′), 72.6 (C-5 or C5′), 72.3 (O-CH2-Ph), 71.9 (O-CH2-Ph), 63.0 (C-6 or C-6′), 62.2 (C-6 or C-6′), 46.2 (C-7). HRMS (ESI) m/z [M + K]+ calculated for [C61H64O10S2K]+: 1059.357, found: 1059.366.
Dithioacetal 6b. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6b was obtained in 16% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 0/1). + 152 (c 0.9, CHCl3). IR 3428 cm−1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.69 (J = 8.2 Hz, 2H, H-10), 7.62 (J = 8.1 Hz, 2H, H-9), 7.45–7.09 (m, 30H, Ph), 5.93 (d, J = 5.5 Hz, 1H, H-1 or H-1′), 5.06 (s, 1H, H-7), 4.99 (d, J = 10.9 Hz, 1H, O-CH2-Ph), 4.91–4.78 (m, 6H, O-CH2-Ph, H-1 or H-1′), 4.71 (d, J = 10.9 Hz, 1H, O-CH2-Ph), 4.63 (d, J = 11.2 Hz, 1H, O-CH2-Ph), 4.55 (d, J = 10.5 Hz, 2H, O-CH2-Ph), 4.43 (d, J = 11.8 Hz, 1H, O-CH2-Ph), 4.39 (d, J = 11.7 Hz, 1H, O-CH2-Ph), 4.17 (ddd, J = 9.9, 7.4, 2.0 Hz, 1H, H-5 or H-5′), 4.09 (ddd, J = 9.7, 6.9, 2.8 Hz, 1H, H-5 or H-5′), 3.97–3.76 (m, 5H, H-6, H-6′, H-3, H-3′, H-2 or H-2′) 3.68 (dd, J = 9.4, 5.6 Hz, 1H, H-2 or H-2′), 3.64–3.54 (m, 2H, H-6, H-6′) 3.35 (dd, J = 10.0; 8.9 Hz, 1H, H-4 or H-4′), 3.28 (dd, J = 10.0; 8.8 Hz, 1H, H-4 or H-4′). 13C NMR (100 MHz, CDCl3) δ (ppm) 143.7 (C-8), 138.55 (Cq-Ar), 138.5 (Cq-Ar), 138.0 (Cq-Ar), 137.7 (Cq-Ar), 137.5 (Cq-Ar), 137.2 (Cq-Ar), 128.8-127.8 (34 × CHAr), 125.9 (C9, q, J C-F = 4 Hz), 82.65 (CH) 82.6 (CH), 82.55 (CH), 82.0 (CH, C-1 or C-1′), 79.4 (CH), 79.0 (C-2 or C-2′), 78.4 (C-4 or C-4′), 77.7 (C-4 or C-4′), 75.9 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.2 (O-CH2-Ph), 72.85 (C-5 or C-5′), 72.8 (C-5 or C-5′), 72.5 (O-CH2-Ph), 72.4 (O-CH2-Ph), 63.1 (C-6 or C-6), 62.1 (C-6 or C-6′), 45.2 (C-7). 19F NMR (376 MHz, CDCl3) δ (ppm) −66.5 (3F, s, CF3). HRMS (ESI) m/z [M + K]+ calculated for [C62H63F1O10S2K]+: 1127.345, found: 1127.332.
Dithioacetal 6c. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6c was obtained in 45% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 0/1). + 151 (c 1.1, CHCl3). IR 3437 cm−1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.49 (d, J = 8.8 Hz, 2H, H-10), 7.44–7.37 (m, 2H, Ph), 7.37–7.19 (m, 26H, Ph), 7.18–7.11 (m, 2H, Ph), 6.88 (J = 8.8 Hz, 2H, H-9), 5.93 (d, J = 5.4 Hz, 1H, H-1 or H-1′), 5.01–4.95 (m, 3H, H-7, H-1 or H-1′, O-CH2-Ph), 4.90–4.63 (m, 3H, O-CH2-Ph), 4.81 (d, J = 5.0 Hz, 2H, O-CH2-Ph), 4.78 (d, J = 4.9 Hz, 1H, O-CH2-Ph), 4.69 (d, J = 10.8 Hz, 1H, O-CH2-Ph), 4.60 (d, J = 11.4 Hz, 1H, O-CH2-Ph), 4.55 (d, J = 5.4 Hz, 1H, O-CH2-Ph), 4.53 (d, J = 5.1 Hz, 1H, O-CH2-Ph), 4.39 (d, J = 11.5 Hz, 1H, O-CH2-Ph), 4.32 (d, J = 11.6 Hz, 1H, O-CH2-Ph), 4.20 (ddd, J = 10.1, 7.4, 2.5 Hz, 1H, H-5 or H-5′), 4.13 (ddd, J = 9.7, 6.9, 2.8 Hz, 1H, H-5 or H-5′), 3.96–3.79 (m, 5H, H-6, H-6′, H-3, H-3′, H-2 or H-2′), 3.80 (s, 3H, OCH3), 3.67 (dd, J = 9.5; 5.6 Hz, 1H, H-2 or H-2′), 3.64-3.54 (m, 2H, H-6, H-6′) 3.35 (dd, J = 9.8; 8.9 Hz, 1H, H-4 or H-4′), 3.27 (dd, J = 10.0; 8.9 Hz, 1H, H-4 or H-4′) 13C NMR (100 MHz, CDCl3) δ (ppm) 159.7 (C-11), 138.65 (Cq-Ar), 138.6 (Cq-Ar), 138.1 (Cq-Ar), 137.8 (Cq-Ar), 137.7 (Cq-Ar), 137.4 (Cq-Ar), 131.3 (C-8), 129.5 (C10), 128.7–127.7 (34 x CHAr), 114.2 (C-9), 82.75 (CH) 82.7 (CH), 82.6 (CH), 81.9 (C-1 or C-1′), 79.4 (CH), 78.9 (C-2 or C-2′), 78.4 (C-4 or C-4′), 77.8 (C-4 or C-4′), 75.9 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.1 (O-CH2-Ph), 72.7 (C-5 or C-5′), 72.5 (C-5 or C-5′), 72.2 (O-CH2-Ph), 71.9 (O-CH2-Ph), 63.0 (C-6 or C-6′), 62.1 (C-6 or C-6′), 55.5 (OCH3), 45.7 (C-7). HRMS (ESI) m/z [M + K]+ calculated for [C62H66O11S2K]+: 1089.368, found: 1089.367.
Dithioacetal 6d. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6d was obtained in 30% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 4/6). + 136 (c 1, CHCl3). IR 3440 cm−1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.35–7.26 (m, 30H, Ph), 5.85 (d, J = 4.5 Hz, 1H, H-1 or H-1′), 5.37 (d, J = 5.4 Hz, 1H, H-1 or H-1′), 5.01–4.95 (m, 2H, O-CH2-Ph), 4.93 (s, 1H, O-CH2-Ph), 4.90–4.85 (m, 2H, O-CH2-Ph), 4.84 (s, 1H, O-CH2-Ph), 4.79 (t, J = 11.1 Hz, 2H, O-CH2-Ph), 4.74 (m, 1H, O-CH2-Ph), 4.71–4.70 (m, 2H, O-CH2-Ph), 4.63–4.55 (m, 5H, O-CH2-Ph), 4.16–4.09 (m, 1H, H5 or H-5′), 4.05–3.95 (m, 2H, H-7, H-4 or H5′), 3.91–3.75 (m, 6H, H-6, H-6′, H-3, H-3′, H-2, H2′), 3.68–3.5 (m, 2H, H-6, H6′), 3.47–3.41 (m, 1H, H-4 or H-4′), 3.36 (dd, J = 10.1; 8.4 Hz, 1H, H-4 or H-4′), 1.93–1.87 (m, 2H, H-8), 1.76–1.64 (m, 1H, H-9), 1.50–1.39 (m, 1H, H-9), 0.96–0.92 (m, 3H, H-10). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.8 (Cq-Ar), 138.6 (Cq-Ar), 138.1 (Cq-Ar), 137.8 (Cq-Ar), 137.7 (Cq-Ar), 137.65 (Cq-Ar),128.7-128.0 (30 × CHAr), 82.8 (CH), 82.52 (CH), 82.5 (CH), 81.3 (C-1 or C-1′), 79.6 (CH), 79.3 (CH), 78.4 (C-4 or C4′), 77.4 (C4 or C-4′), 75.9 (CO-CH2-Ph), 75.8 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.4 (O-CH2-Ph), 72.9 (C5 or C5′), 72.5 (O-CH2-Ph), 72.3 (C5 or C5′), 72.2 (O-CH2-Ph), 62.8 (C-6 or C6′), 61.9 (C6 or C-6′), 45.0 (C-7), 38.5 (C-8), 20.5 (C-9), 13.9 (C-10). HRMS (ESI) m/z [M + K]+ calculated for [C59H68O10S2Na]+: 1023.415, found: 1023.420.
Dithioacetal 6e. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6e was obtained in 33% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 0/1). + 173 (c 0.95, CHCl3). IR 3452 cm-1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.45–7.17 (m, 30H, Ph), 5.85 (d, J = 4.3 Hz, 1H, H-1 or H-1′), 5.38 (d, J = 5.3 Hz, 1H, H-1 or H-1′), 5.02–4.54 (m, 12H, O-CH2-Ph), 4.11 (ddd, J = 10.1, 6.8, 2.5 Hz, 1H, H-5 or H-5′), 4.05–3.93 (m, 2H, H-5 or H-5′, H-7), 3.91–3.75 (m, 6H, H-6, H-6′, H-3, H-3′, H-2, H2′), 3.66 (dd, J = 12.6; 5.8 Hz, 1H, H-6 or H-6′), 3.59 (dd, J = 11.6, 6.6 Hz, 1H, H-6 or H-6′), 3.48–3.42 (m, 1H, H-4 or H-4′), 3.36 (dd, J = 9.9; 8.6 Hz, 1H, H-4 or H-4′), 1.96–1.88 (m, 2H, H-8), 1.76–1.61 (m, 1H, H-9), 1.46–1.36 (m, 1H, H-9), 1.36–1.25 (m, 4H, H-10, H11), 0.97–0.85 (m, 3H, H-12). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.8 (Cq-Ar), 138.6 (Cq-Ar), 138.1 (Cq-Ar), 137.8 (Cq-Ar), 137.7 (Cq-Ar), 137.6 (Cq-Ar), 128.7-127.7 (30 × CHAr), 82.8 (CH), 82.55 (CH), 82.5 (CH), 81.4 (C-1 or C-1′), 79.5 (CH), 79.3 (CH), 78.4 (C4 or C-4′), 77.4 (C-4 or C4′), 75.9 (O-CH2-Ph), 75.8 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.4 (O-CH2-Ph), 72.8 (O-CH2-Ph), 72.5 (C-5 or C-5′), 72.3 (C-5 or C-5′), 72.25 (O-CH2-Ph), 62.8 (C-6 or C-6′), 61.9 (C-6 or C-6′), 45.4 (C-7), 36.3 (C-8), 31.7 (C-10 or C-11), 26.7 (C-9), 22.7 (C-10 or C-11), 14.2 (C-12). HRMS (ESI) m/z [M + K]+ calculated for [C60H70O10S2K]+: 1053.404, found: 1053.391.
Dithioacetal 6f. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6f was obtained as a solid in 25% yield after flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 4/6. + 128 (c 1.3, CHCl3). IR 3445 cm−1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.39–7.29 (m, 30H, Ph), 5.76 (d, J = 4.8 Hz, 1H, H-1 or H-1′), 5.38 (d, J = 5.4 Hz, 1H, H-1 or H-1′), 5.01–4.66 (m, 9H, O-CH2-Ph), 4.61 (m, 2H, O-CH2-Ph), 4.63–4.56 (m, 1H, O-CH2-Ph), 4.10–4.05 (m, 1H, H-5 or H-5′), 4.03–3.98 (m, 1H, H-5 or H-5′), 3.90–3.77 (m, 6H, H-7, H-3, H-3′, H6, H6′, H-2 or H-2′), 3.76 (dd, J = 9.5; 5.4 Hz, 1H, H-2 or H-2′), 3.66 (dd, J = 12.3; 5.9 Hz, 1H, H-6 or H-6′), 3.59 (dd, J = 11.6, 6.2 Hz, 1H, H-6 or H-6′), 3.44 (t, J = 9.3 Hz, 1H, H-4 or H-4′), 3.36 (t, J = 9.2 Hz, 1H, H-4 or H-4′), 1.19 (s, 9H, H-9). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.9 (Cq-Ar), 138.5 (Cq-Ar), 138.0 (Cq-Ar), 137.9 (Cq-Ar), 137.6 (Cq-Ar), 137.5 (Cq-Ar), 128.7-127.7, (30xCHAr), 86.0 (CH, C-1′), 83.0 (CH), 82.0 (CH), 81.3 (CH, C-1), 79.8 (C2′), 79.0 (CH), 78.4 (C4), 77.7 (C4′) 75.8 (O-CH2-Ph), 75.6 (O-CH2-Ph), 75.4 (O-CH2-Ph), 73.8 (O-CH2-Ph), 73.0 (C-5), 72.2 (O-CH2-Ph), 72.1 (C-5) 62.8 (C-6′), 62.2 (C-6), 59.6 (C-7), 38.6 (C-8), 28.3 (C-9). HRMS (ESI) m/z [M + Na]+ calculated for [C58H66O10S2Na]+: 1009.399, found: 1009.404.
Dithioacetal 6g. According to the general procedure for the synthesis of diglycoside thioketals 6, compound 6g was obtained in 50% yield after flash column chromatography (Petroleum Ether/EtOAc, 9/1 to 1/9). + 388 (c 0.51, CHCl3). IR 3425 cm−1 (broad O-H). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.53–7.51 (m, 2H, Ph), 7.34–7.26 (m, 3H, Ph), 5.98–5.82 (m, 6H, 5x OCH2CH=CH2, H1 or H-1′), 5.70–5.61 (m, 1H, OCH2CH=CH2), 5.38–5.06 (m, 12H, 6xOCH2CH=CH2), 5.02–4.96 (m, 3H, H1′, H7, 1 CH-O), 4.38–4.04 (m, 13H, 6 × OCH2CHCH2), 3.98 (dd, J = 11.4, 2.7 Hz 1H, H-6 or H-6′), 3.88 (dd, J = 11.7, 2.7 Hz, 1H, H-6 or H-6′), 3.78–3.47 (m, 8H, H6, H6′, 6H CH-O), 3.19 (dd, J = 9.7, 9.6 Hz, 1H, H-4 or H-4′), 3.10 (dd, J = 9.5, 9.6 Hz, 1H, H-4 or H-4′). 13C NMR (100 MHz, CDCl3) δ (ppm) 139.3 (Cq-Ar) 135.3, 135.2, 134.7, 134.5, 134.4, 134.3, (6x OCH2CH=CH2), 128.7 (2 × CHAr), 128.4, (CHAr), 128.3, (2 × CHAr), 118.0, 117.8, 117.7, 117.2, 116.9, 116.8 (6 × OCH2CH=CH2), 82.8 (C-1 or C-1′), 82.2, 82.1, 81.9, 79.05, 79.0 (5xCH), 78.3 (C-4 or C-4′), 77.7 (C4 or C4′), 74.5 (2 × OCH2CH=CH2), 74.4 (OCH2CH=CH2), 74.0 (OCH2CH=CH2), 72.6 (CH), 72.5 (CH), 71.3 (OCH2CH=CH2), 71.1 (OCH2CH=CH2), 63.2 (C-6 or C-6′), 62.2 (C-6 or C-6′), 46.1 (C-7). HRMS (ESI) m/z [M + Na]+ calculalted for [C37H52NaO10S2]+: 743.290, found 743.290.
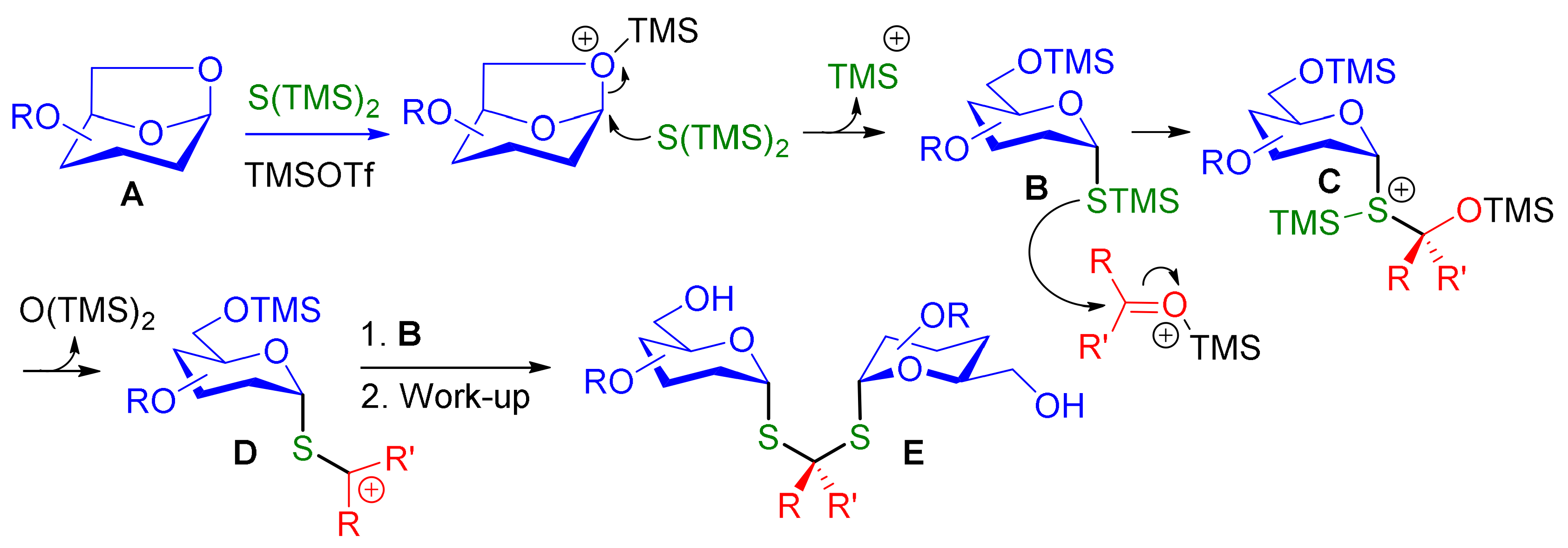
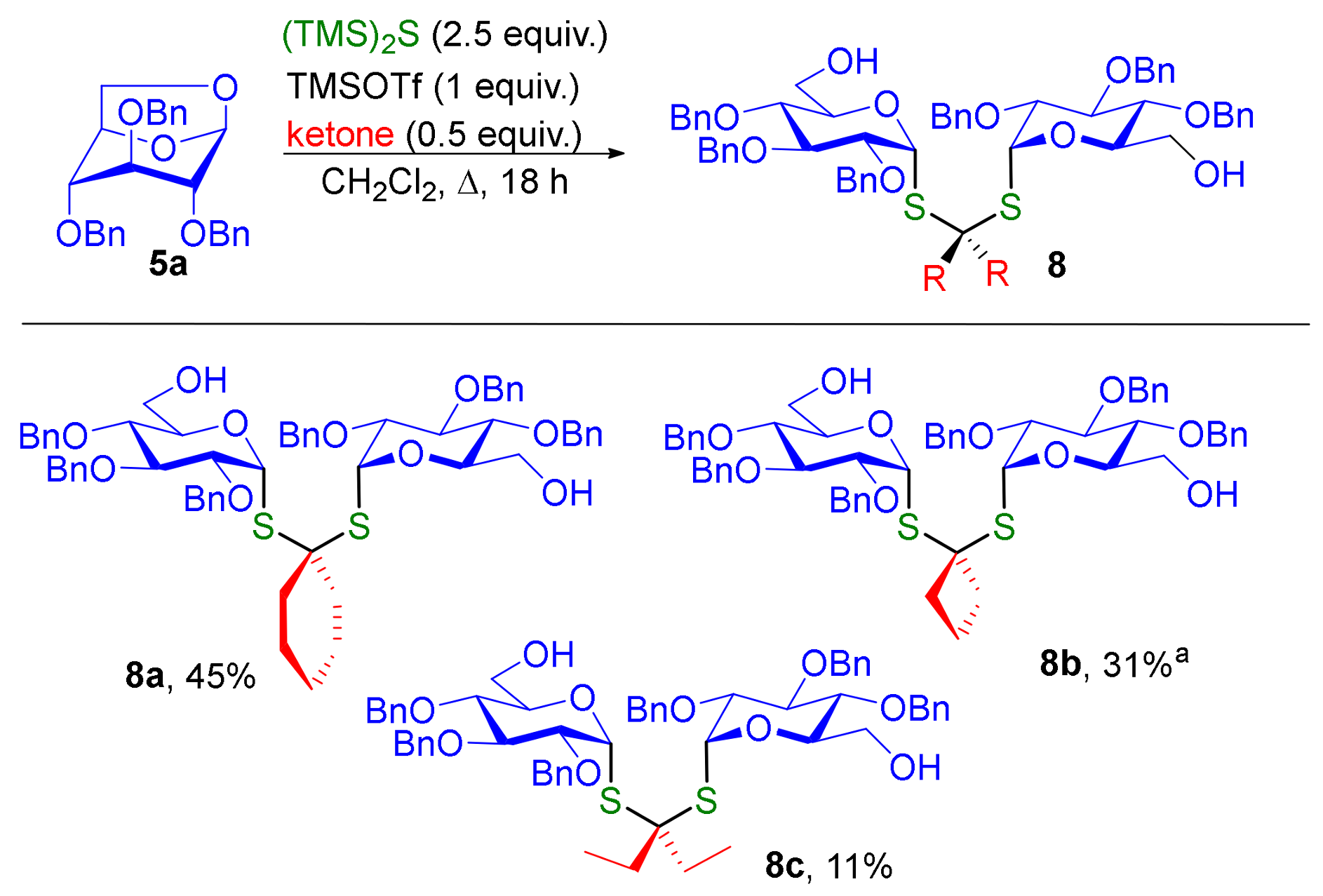
3.3. Synthesis of Dithioketal-α,α-Diglycosides 8 and Characterization of Compound 3
Diglycoside thioketal 8a. In a tube, to a solution of cyclohexanone (1 equiv., 13.2 mg, 0.014 mL, 0.134 mmol) and anhydroglucose 5a (2 equiv., 116 mg, 0.269 mmol) in CH2Cl2 (0.6 mL) was added (TMS)2S (5 equiv., 0.126 mL, 0.672 mmol) and TMSOTf (2 equiv., 0.0488 mL, 0.269 mmol). The tube was sealed and the mixture was heated at 60 °C for 18 h. The mixture was washed with saturated aqueous NaHCO3 (40 mL) and extracted with CH2Cl2 (3 × 50 mL). The organics layers were combined and washed with brine (40 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude obtained was purified by flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 1/1) to afford compound 8a (61 mg) in 45% yield. + 114 (c 1, CH2Cl2). IR 3463 (O-H, weak, broad), 2928 (C-H) cm-1. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.31–7.13 (m, 30H, H-Ar), 5.79 (d, J = 5.4 Hz, 2H, H-1), 4.84–4.48 (m, 12H, Ph-CH2-O), 4.06 (dt, J = 10.1, 3.1 Hz, 2H, H-5), 3.73 (t, J = 9.5 Hz, 2H, H-3), 3.69 (m, 4H, H-6), 3.60 (dd, J = 9.8, 5.6 Hz, 2H, H-2), 3.46 (t, J = 9.3 Hz, 2H, H-4), 1.96 (t, J = 5.6 Hz, 4H, H-8), 1.54 (m, 4H, H-9), 1.31 (m, 2H, H-10). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.5 (Cq-Ar,) 138.2 (Cq-Ar,), 138.1 (Cq-Ar) 128.64, 128.60, 128.54, 128.51, 128.18, 128.14, 128.09, 128.05, 127.84, 127.77, 127.67 (CH Ar), 83.4 (C-3), 82.6 (C-1), 79.6 (C-2), 77.4 (C-4), 75.7 (Ph-CH2-O), 75.3 (Ph-CH2-O), 73.0 (Ph-CH2-O), 72.7 (C-5), 64.1 (C-7), 62.0 (C-6), 39.9 (C-8), 25.3 (C-10), 23.1 (C-9). HRMS (ESI) m/z [M + Na]+ calculated for [C60H68O10S2Na]+: 1035.415; found 1035.416.
Diglycoside thioketal 8b. In a tube, to a solution of anhydroglucose 5a (2 equiv., 100 mg, 0.231 mmol) in CH2Cl2 (0.6 mL) was added (TMS)2S (2.8 equiv., 0.0608 mL, 0.324 mmol) and TMSOTf (2.2 equiv., 0.0461 mL, 0.254 mmol). The tube was sealed and the mixture was heated at 50 °C for 2 h. Then cyclobutanone (1 equiv., 8.1 mg, 0.00871 mL, 0.116 mmol) was added to the mixture at −70 °C and stirred 15 h. The mixture was washed with saturated aqueous NaHCO3 (40 mL) and extracted with CH2Cl2 (3 × 50 mL). The organics layers were combined and washed with brine (40 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude obtained was purified by flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 0/1) to afford compound 8b (35 mg) in 31% yield. + 87 (c 1, CH2Cl2). IR 3466 (O-H, weak, broad), 2925 (C-H) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.31–7.13 (m, 30H, H-Ar), 5.74 (d, J = 5.6 Hz, 2H, H-1), 4.79 (t, J = 10.9 Hz, 4H, O-CH2-Ph), 4.69 (d, J = 10.4 Hz, 2H, O-CH2-Ph) 4.57 (d, J = 10.9 Hz, 2H, O-CH2-Ph), 4.09 (dt, J = 9.7, 3.1 Hz, 2H, H-5), 3.79–3.66 (m, 6H, H-3, H-6), 3.60 (dd, J = 9.9, 5.7 Hz, 2H, H-2), 3.47 (dd, J = 9.6, 9.0 Hz, 2H, H-4), 2.58–2.47 (m, 2H, H-8a), 2.40–2.31 (m, 2H, H-8b), 2.06 (q, J = 7.6 Hz, 2H, H-9). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.4, (Cq-Ar), 138.1 (Cq-Ar), 137.9 (Cq-Ar), 128.5, 128.4, 128.03, 127.97, 127.93, 127.7, 127.3 (CHAr), 83.8 (C-1), 83.2 (C-3), 79.4 (C-2), 77.3 (C-4), 75.6 (O-CH2-Ph), 75.1 (O-CH2-Ph), 72.7 (O-CH2-Ph), 72.5 (C-5), 61.8 (C-6), 59.1 (C-7), 38.9 (C-8), 18.0 (C-9). HRMS (ESI) m/z [M + Na]+ calculated for [C58H64O10S2 Na]+: 1007.383; found 1007.382.
Diglycoside thioketal 8c. In a tube, to a solution of 3-pentanone (1 equiv., 9.96 mg, 0.0122 mL, 0.116 mmol) and anhydroglucose 5a (2 equiv., 100 mg, 0.231 mmol) in CH2Cl2 (0.6 mL) was added (TMS)2S (5 equiv., 103 mg, 0.109 mL, 0.578 mmol) and TMSOTf (2 equiv., 51.4 mg, 0.0419 mL, 0.231 mmol). The tube was sealed and the mixture was heated at 50 °C for 2 h. The mixture was washed with saturated aqueous NaHCO3 (40 mL) and extracted with CH2Cl2 (3 × 50 mL). The organics layers were combined and washed with brine (40 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude obtained was purified by flash column chromatography (Petroleum Ether/EtOAc, 8/2 to 4/6) to afford compound 8c (13 mg) in 11% yield. + 104 (c 1, CH2Cl2). IR 3465 (O-H, weak, broad), 2875 (C-H) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.40–7.15 (m, 30H, H-Ar), 5.83 (d, J = 5.7 Hz, 2H, H-1), 4.87 (d, J = 6.3 Hz, 2H, O-CH2-Ph), 4.85 (d, J = 6.2 Hz, 2H, O-CH2-Ph), 4.76 (d, J = 10.9 Hz, 2H, O-CH2-Ph), 4.66–4.56 (m, 6H, O-CH2-Ph), 4.10 (dt, J = 9.8, 3.2 Hz, 2H, H-5), 3.84–3.75 (m, 6H, H-3, H-6), 3.63 (dd, J = 9.9, 5.6 Hz, 2H, H-2), 3.52 (dd, J = 10.0, 8.9 Hz, 2H, H-4), 2.02–1.92 (m, 2H, H-8a), 1.92–1.81 (m, 2H, H-8b), 0.99 (t, J = 7.3 Hz, 6H, H-9). 13C NMR (100 MHz, CDCl3) δ (ppm) 138.5 (Cq-Ar), 138.2 (Cq-Ar), 138.1 (Cq-Ar) 128.64, 128.53, 128.50, 128.17, 128.06, 127.82, 127.76, 127.5 (CH Ar), 83.3 (C-3), 83.9 (C-1), 79.6 (C-2), 77.4 (C-4), 75.7 (Ph-CH2-O), 75.3, (Ph-CH2-O), 73.1 (Ph-CH2-O), 72.8 (C-5), 68.5 (C-7), 61.9 (C-6), 31.9 (C-8), 9.1 (C-9). HRMS (ESI) [M + K]+ calculated for [C59H68O10S2K]+: 1039.389; found 1039.394.
Diglycoside thioketal 3. + 128 (c 1, CH2Cl2). IR 2174 (C≡C, weak, sharp) cm−1. 1H NMR (400 MHz, CDCl3) δ (ppm) 5.56 (d, J = 5.7 Hz, 2H, H-1), 4.66–4.24 (m, 12H, H-9), 3.97 (dd, J = 9.9 and 5.7 Hz, 2H, H-2), 3.91–3.80 (m, 4H, H-5, H-6a), 3.73 (dd, J = 11.8, 5.2 Hz, 2H, H-6b), 3.63 (t, J = 9.2 Hz, 2H, H-3), 3.43 (t, J = 9.4 Hz, 2H, H-4), 1.68 (s, 6H, H-8), 1.12–0.98 (m, 126H, H-TIPS). 13C NMR (100 MHz, CDCl3) δ (ppm) 103.9, 103.7, 102.9 (3 × C-10), 88.9, 87.8, 87.5 (3 × C-11), 83.6 (C-1), 82.7 (C-3), 77.2 (C-2), 76.7 (C-4), 73.1 (C-5), 62.5 (C-6), 61.0, 60.7, 59.1 (3 × C-9), 58.7 (C-7), 32.9 (C-8), 18.7 (CH3-CH-Si), 11.3 (CH3-CH-Si). HRMS (ESI) m/z [M + Na]+ calculated for [C87H160O10S2Si6Na]+: 1619.996; found 1619.989.

 and
and 




{kind=link}
{kind=link}
{kind=link}
{kind=link}

