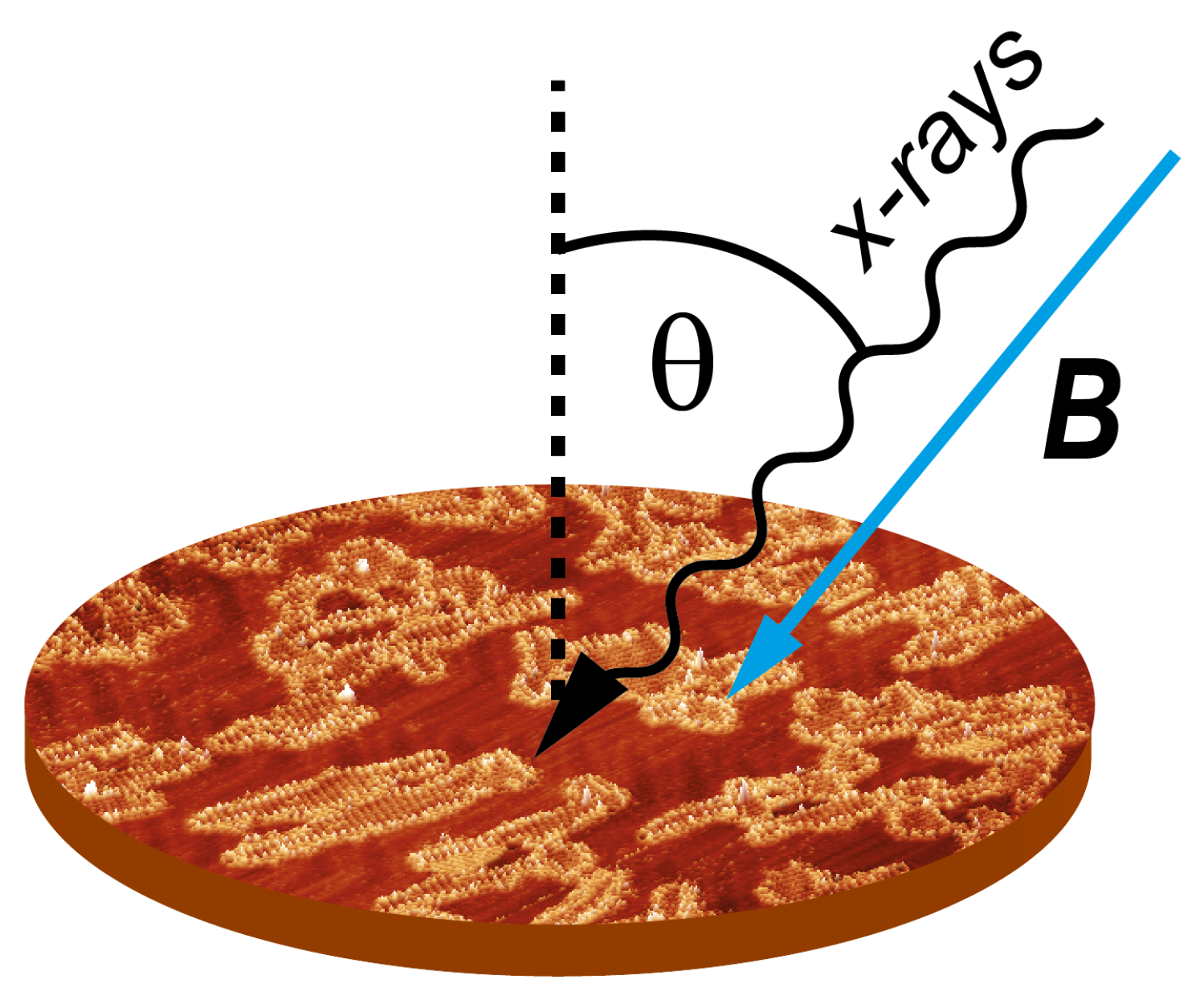
3.1. X-ray Magnetic Circular Dichroism Data
The XMCD intensity variation as a function of the applied magnetic field (
B) defines a curve that is proportional to the system magnetization. Therefore, when the value of the spin magnetic moments at the metal centers (
S) , the temperature (
T), and the Landé
g-factor are known, one can use simple models to simulate the magnetization response. A good reference to be considered is the case of paramagnetic behavior (spins responding individually to the applied magnetic field) that can be represented by a Brillouin function. Whenever a preference for ferromagnetic (FM) or antiferromagnetic (AFM) coupling between spins appears, the corresponding magnetization curves will show higher or lower curvature, respectively, than the corresponding Brillouin function for the same
S,
T, and
g-factor values. In this way, in principle, one can decide about the type of magnetic coupling between localized spins at the metal centers, as long as the value of the spin (
S) is known. Note that, in the presence of strong magnetic anisotropies and high orbital angular moments, the analysis becomes more involved [
28]. However, here we can follow this simplified scheme, as shown below. According to our DFT calculations, described in
Section 3.3, Mn atoms in Mn–TCNQ have a localized spin magnetic moment close to
, although somewhat lower, while Ni atoms in Ni–TCNQ have a much lower spin close to
, although somewhat higher. Therefore, we use the values
and
for Mn and Ni, respectively, to perform our XMCD analysis that includes fitting curves to XMCD data based on Weiss mean-field theory described in the next section, where
J and
D are defined, and also a comparison with the corresponding Brillouin functions.
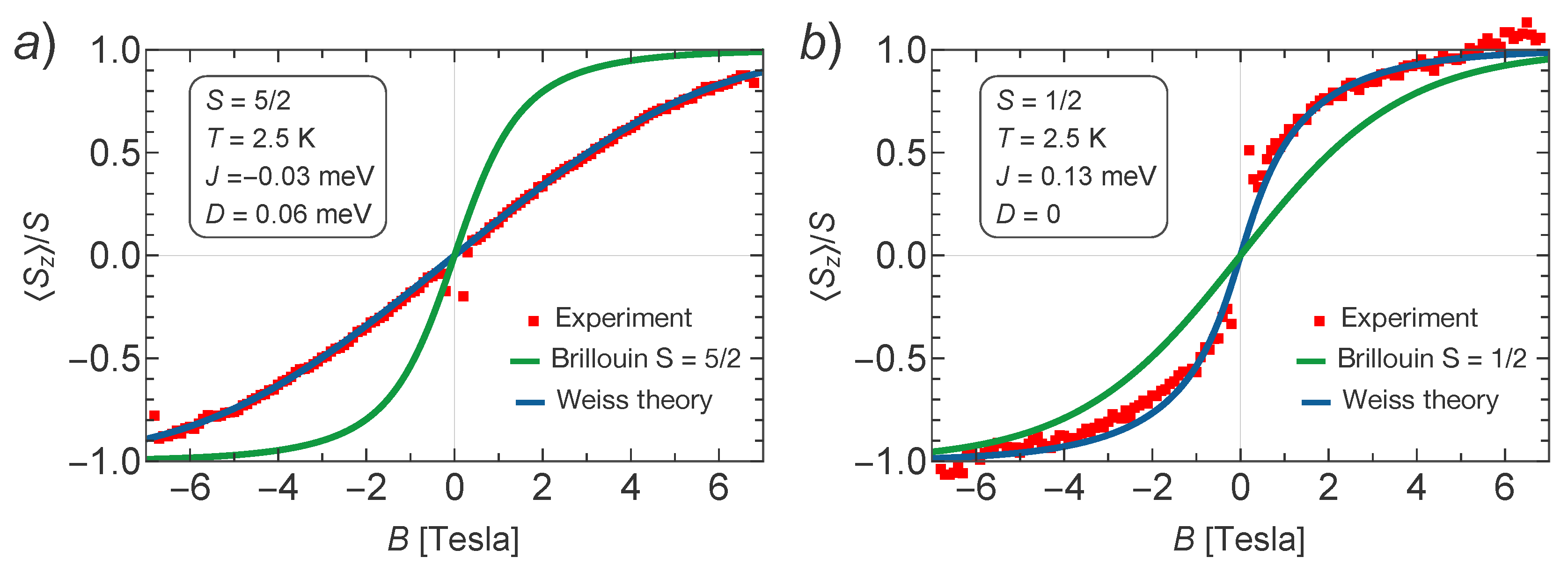
The results are shown in
Figure 3a,b for Mn–TCNQ and Ni–TCNQ, respectively. It is evident that in Mn–TCNQ the coupling between Mn spins is AFM, while in Ni–TCNQ it is FM. Additionally, the fitted values of the exchange coupling constants reveal a weaker coupling between Mn spins (
meV) as compared to the coupling between Ni spins (
meV), while the single ion anisotropy parameter
meV corresponds to a weak anisotropy with in-plane magnetization for Mn–TNCQ and
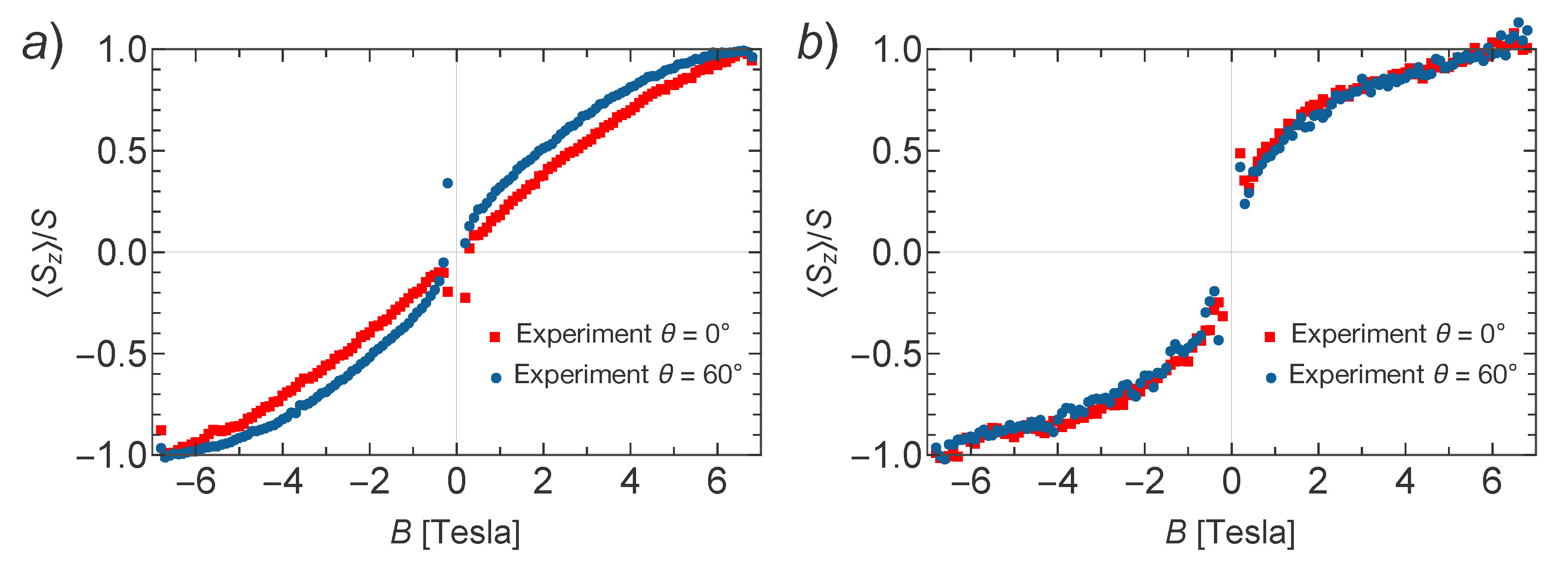
to the absence of anisotropy for Ni–TCNQ. In order to learn more about the magnetic anisotropy of these systems, in
Figure 4 we plot a comparison of XMCD data obtained for perpendicular and grazing incidence for Mn–TCNQ and Ni–TCNQ, the former showing a mild angular dependence with stronger intensity for grazing incidence, i.e., a fingerprint of magnetic anisotropy in the system with in-plane magnetization. Incidentally, this weak anisotropy is only observed at low temperatures. However, in the Ni–TCNQ XMCD data there is no significant angular dependence, which means a negligible magnetic anisotropy. A value of the Ni atom spin
corresponds to the absence of single ion anisotropy [
29].
3.2. Model for Mn–TCNQ and Ni–TCNQ
In Mn–TCNQ, the coupling between local moments is antiferromagnetic and occurs by means of the Anderson superexchange mechanism [
15,
30]. In perturbation theory, the superexchange interaction was found to be dominated by a virtual process in which two electrons hop from the lowest unoccupied molecular orbital (LUMO) of the TCNQ molecule, which is doubly occupied in this MOCN, onto two adjacent Mn atoms [
11]. Inclusion of additional molecular orbitals, such as the highest occupied molecular orbital (HOMO), leads to a generic superexchange interaction with coupling constants
,
, and
, as shown in
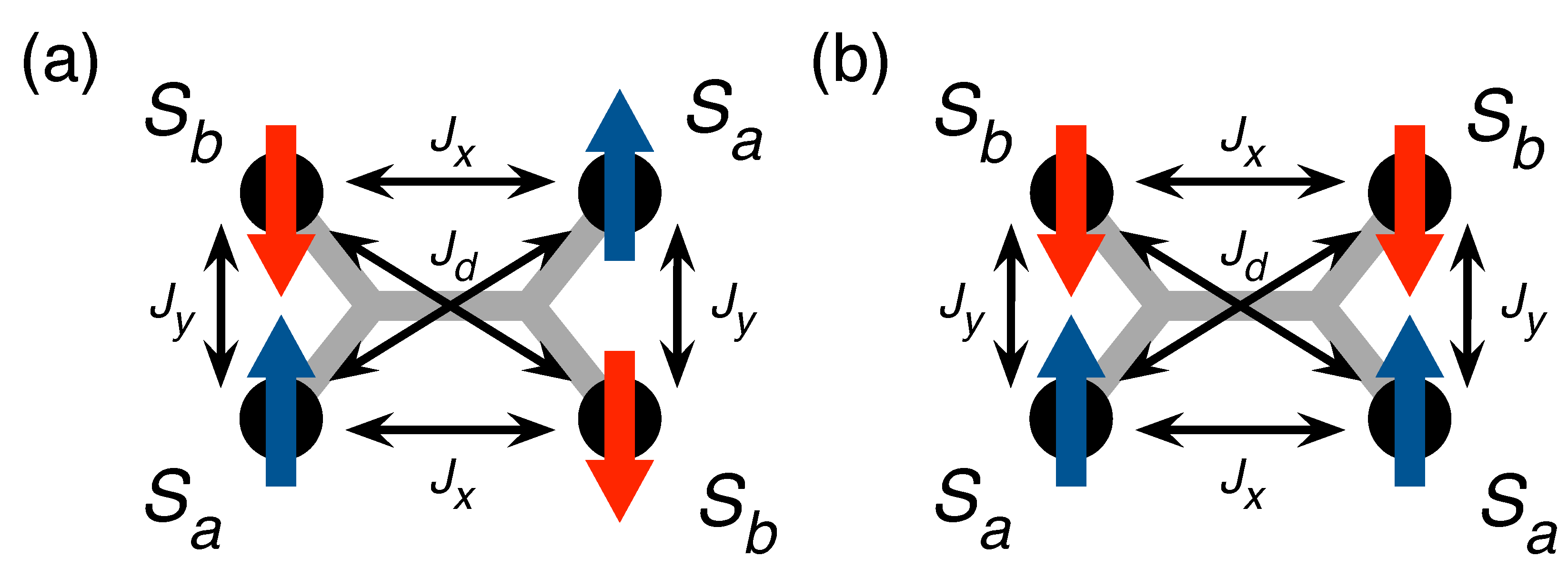
Figure 5. The model Hamiltonian describing the magnetic properties of Mn–TCNQ, thus, reads
where
denotes the local moment of the Mn atom (
) on site
i,
D is the single-ion anisotropy energy,
g is the Landé
g-factor (
), and
is the magnetic field. The Heisenberg exchange constant
is restricted to the nearest (
and
) and next-to-nearest (
) neighbors on the rectangular lattice. The summation in the Heisenberg interaction term accounts twice for each pair of interacting sites; hence the presence of the factor
in Equation (
1).
A quick insight into the tendency to order the spins in this model is granted by the Fourier transform of the exchange coupling
,
where
is the two-dimensional wave vector and
is the position of the Mn atom on site
i. For ferromagnetic couplings (
), the maximum of
occurs at
, which indicates that the spin order could be uniform from a mean-field point of view, not addressing the question about its stability against fluctuations in two dimensions. Additional terms, such as the single-ion anisotropy or the Zeeman interaction, may stabilize the uniform spin order.
In contrast, for antiferromagnetic couplings (
), the maximum of
occurs usually at the edge of the Brillouin zone, indicating that the magnetization is staggered in some way over the unit cell. When only nearest neighbors are coupled (
and
), the maxima lie at
and its equivalent points, which results in the usual checkerboard-like antiferromagnetic order (see
Figure 5a). As the diagonal coupling is turned on (assuming an antiferromagnetic
), for a sufficiently large magnitude of
there is a transition from the checkerboard pattern to a so-called superantiferromagnetic state of antiferromagnetically ordered rows or columns. For
, by requiring
at
, we find at
the transition point for antiferromagnetic column formation (see
Figure 5b).
The effect of the diagonal coupling
consists in introducing magnetic frustration [
30,
31] in the spin lattice. We remark here that the special point
is realized to a good approximation in our Mn–TCNQ lattice, because (1) the LUMO of the TCNQ molecule has a weak overlap with the d
and d
orbitals of the Mn atom, as will be shown in the next section, thus, dominating the superexchange, and (2) the direct coupling between the LUMOs of neighboring TCNQ molecules is rather weak. The latter makes it possible to consider two independent paths of superexchange for the nearest neighbors, with each path going separately via one of the two TCNQ molecules connecting the two neighboring Mn atoms. For the diagonal coupling, only one path is possible, which leads to a reduction of the diagonal coupling by a factor of 2 as compared to the nearest-neighbor coupling. With approximations (1) and (2), the coupling constants obey
(see [
11] for further details).
Despite the fact that the Mn–TCNQ lattice may well be in a frustrated magnetic state consisting of a mixture of the two phases in
Figure 5, the XMCD data appear to be consistent with a much simpler description of the magnetization as a function of the
B-field, which is derived from the Weiss mean-field theory, and it faithfully captures weak deviations from the paramagnetic state. The superexchange couplings are rather weak [
11], of the order of
, and the Zeeman term soon dominates. Additionally, there exists a fair amount of single-ion anisotropy, described by the
term in Equation (
1).
We make the mean-field approximation for the model in Equation (
1),
where
gives the local description of the interacting system in terms of the Weiss fields
. The spin averages
can be regarded as variational parameters of the theory. The last term in the first line of Equation (
3) compensates for the double counting of interaction energy occurring in the local Hamiltonian
and plays an important role when calculating the free energy of the interacting system. The minimization of the free energy allows us to determine the values of the order parameters
. The procedure is described in the
Appendix A.
Next, we focus on the XMCD data taken at normal incidence (
), for which the magnetic field is applied along the
-axis,
. For the (checkerboard) antiferromagnetic phase, we use two order parameters
and
, which represent the
-components of the spins in the unit cell as shown in
Figure 5a, and minimize the upper bound to the free energy [
] with respect to the order parameters
and
. Alternatively, one can require stationarity of free energy,
and
, which yields two coupled equations,
where
is the free energy of a single isolated spin. The mean-field solution is obtained from these self-consistent equations. As a rule, several solutions are found. The choice of the physical solution relies again on the lowest value of the free energy. For the superantiferromagnetic phase, we use again two order parameters,
and
, but now they are distributed in the unit cell as shown in
Figure 5b. The mean-field approximation takes into account only the connections (i.e., bonds) between the spins on a local scale, whereas the constrains related to the dimensionality of the systems go unaccounted for. We can, therefore, adapt here all the results derived for the phase in
Figure 5a by simultaneously replacing
and
in all expressions as
The factors 2 and appear here because each connector counts as half a bond in the unit cell, whereas each connector counts as a full bond.
We fit the experimental data for normal magnetic fields in
Figure 3 assuming the relation
, which corresponds to the case when a single orbital of the ligand is dominating the superexchange. We reach a good fit to the experimental data for
meV. Our working assumption was that the critical temperature (
) is sufficiently low as to allow application of the Weiss theory, i.e.,
. This means also that the order parameters
and
are never of opposite sign and are, in fact, equal to each other over the full range of applied magnetic fields. Therefore, the experimental data can equally well be fitted by a ferromagnetic mean-field theory with antiferromagnetic coupling constants. To simplify the matter even further, we consider a square lattice with a single coupling constant
J. Effectively, this coupling constant will be related to the previous coupling constants by equating to each other
at
for both models, which immediately yields
. Using the above value, we arrive at
and
for
.
The same effective model derived from a mean field Hamiltonian with
J,
S and
D parameters can be used for Ni–TCNQ, although its relation with the microscopic Hamiltonian described in [
11] is different. In this case, we find a good fit with
and
for
.
3.3. Spin-Polarized DFT+U Calculations
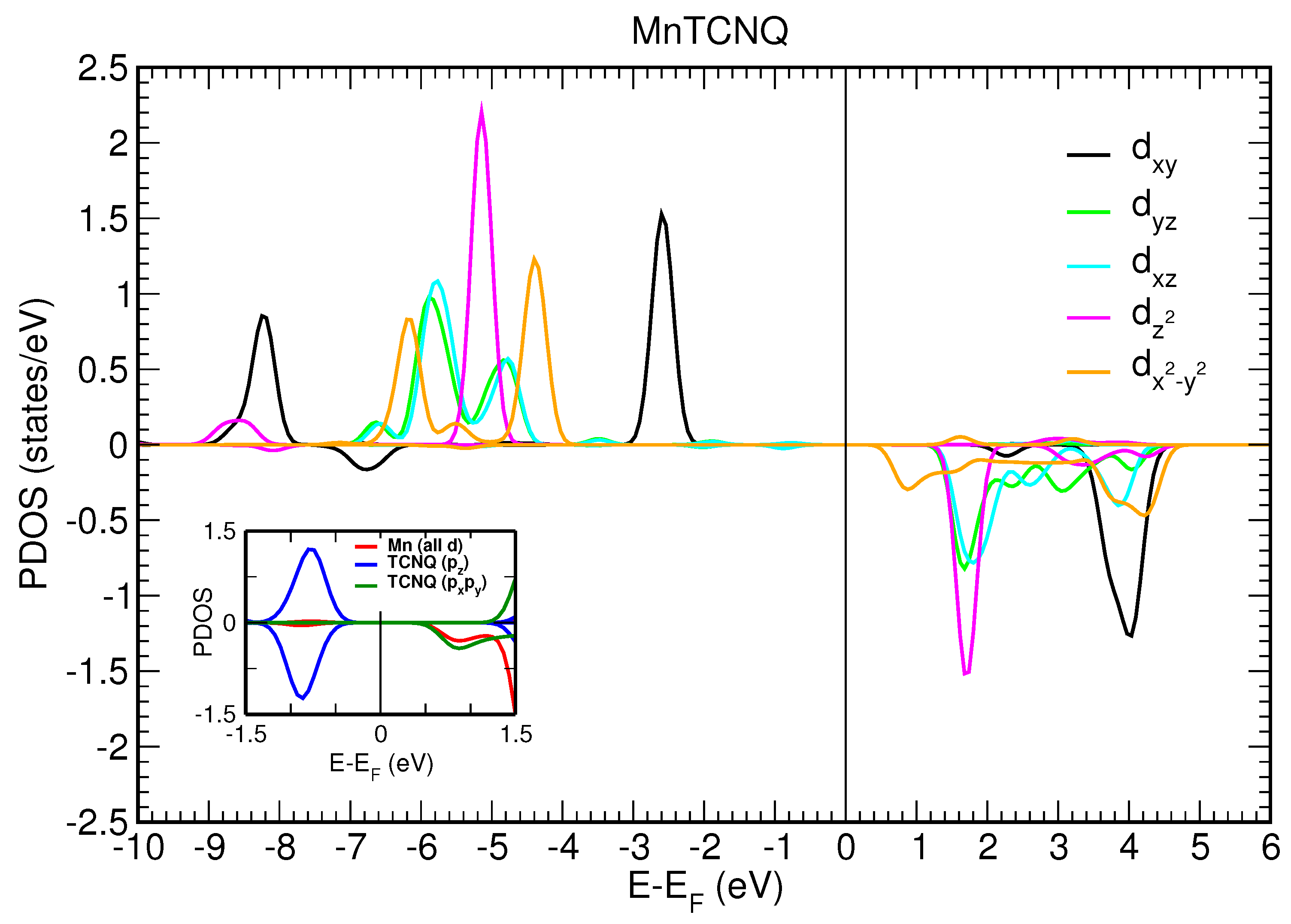
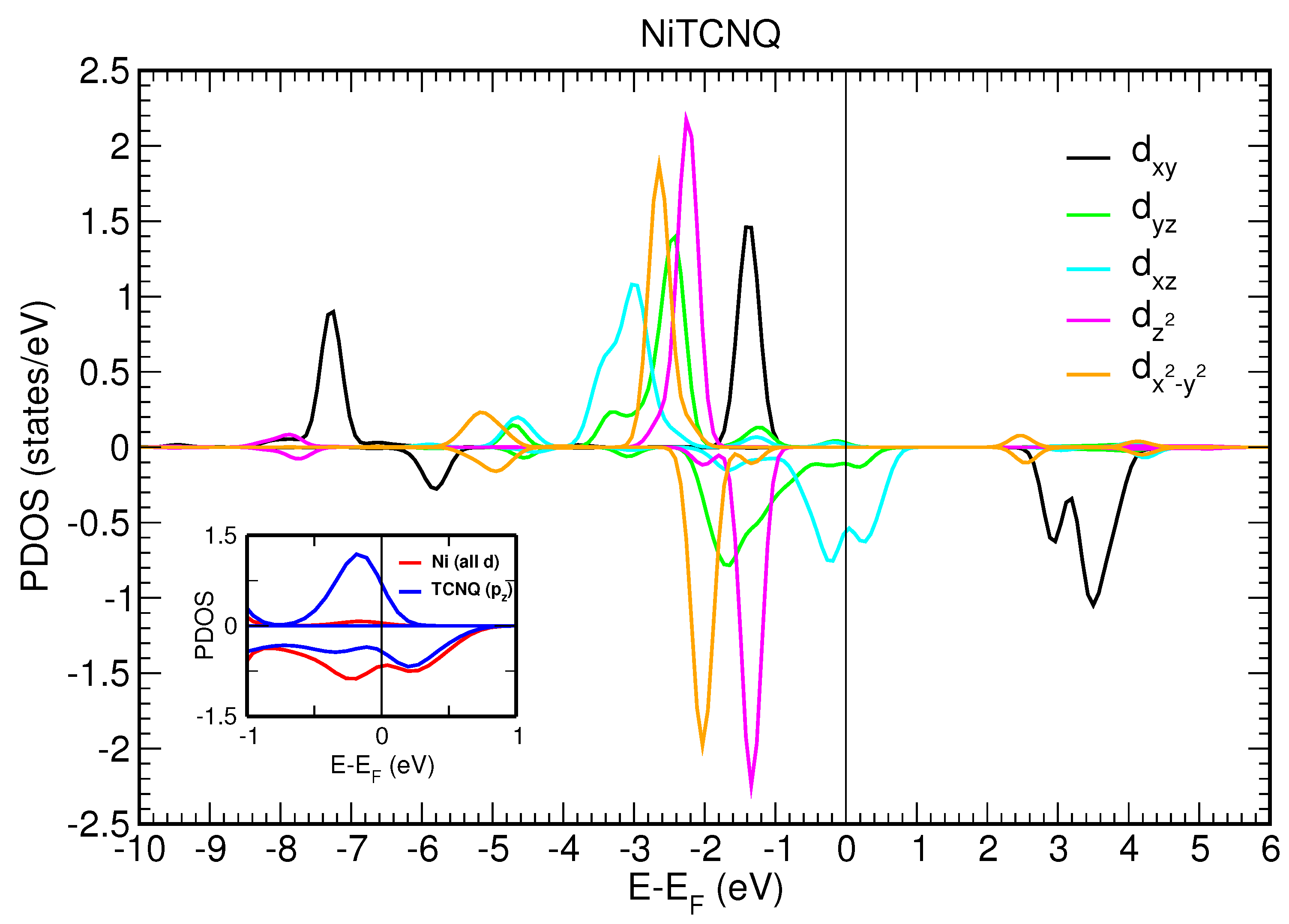
We first consider a two-dimensional free-standing overlayer description for Mn–TCNQ and Ni–TCNQ networks. Both the lattice vectors and atomic positions have been optimized by using an energy minimization procedure within DFT, as described in the Materials and Methods section. The projected densities of states (PDOS) onto different atomic 3d orbitals of the Mn and Ni atoms are shown in
Figure 6 and
Figure 7, respectively. The insets show the PDOS onto atomic p orbitals of the C and N atoms of the organic ligand, as well as onto Mn and Ni 3d states without m number resolution, in a narrow energy range close to the Fermi level. A close inspection of
Figure 6 and
Figure 7 reveals important differences between the two systems under study. The most significant is the half-filling of the 3d states with all the majority spin states occupied in Mn–TNCQ, which corresponds to a value of the spin localized at the Mn atoms approximately equal to
. Meanwhile, in Ni–TCNQ only one minority spin state is fully unoccupied (3d
), which corresponds to a value of the spin localized at the Ni atom of approximately
, although it can be somewhat higher as the minority spin states 3d
and 3d
are partially occupied. Additionally, in Ni–TCNQ, the 3d
and 3d
states are hybridized with TCNQ orbitals close to the Fermi level, in particular the LUMO, giving rise to a delocalized spin density [
11]. This can be seen by comparing the PDOS onto atomic p orbitals of the C and N atoms of the TCNQ organic ligand shown in the insets of
Figure 6 and
Figure 7 for Mn–TCNQ and Ni–TCNQ, respectively. In Ni–TCNQ the LUMO orbital is spin-polarized but this is not the case in Mn–TCNQ, for which the TCNQ LUMO practically does not hybridize with Mn states and is fully occupied. There is another important difference between Ni–TCNQ and Mn–TCNQ: the former is metallic while the second is not. Indeed, the calculated band gap in Mn–TCNQ is rather large (several eV) and translates into large energy barriers for the injection of holes or electrons. As a consequence, electronic charge transfer from the Au(111) surface is expected to play a role in Ni–TCNQ but not in Mn–TCNQ.
Next, using these two optimized structures calculated with a
surface unit cell within the DFT+U method with spin polarization as a starting point, we proceed to double the size of the surface unit cell into a
cell that contains two metal centers (Mn or Ni atoms) and two TCNQ molecules. In this way, we can decide which is the most favorable type of magnetic coupling (ferro- or antiferro-magnetic) between spins localized at the Mn or Ni centers by comparing the values of the corresponding total energies. We consider a checkerboard configuration using oblique vectors in the
surface unit cell and confirm that ferromagnetic coupling is favorable in Ni–TCNQ networks, while in Mn–TCNQ networks antiferromagnetic coupling is preferred in agreement with [
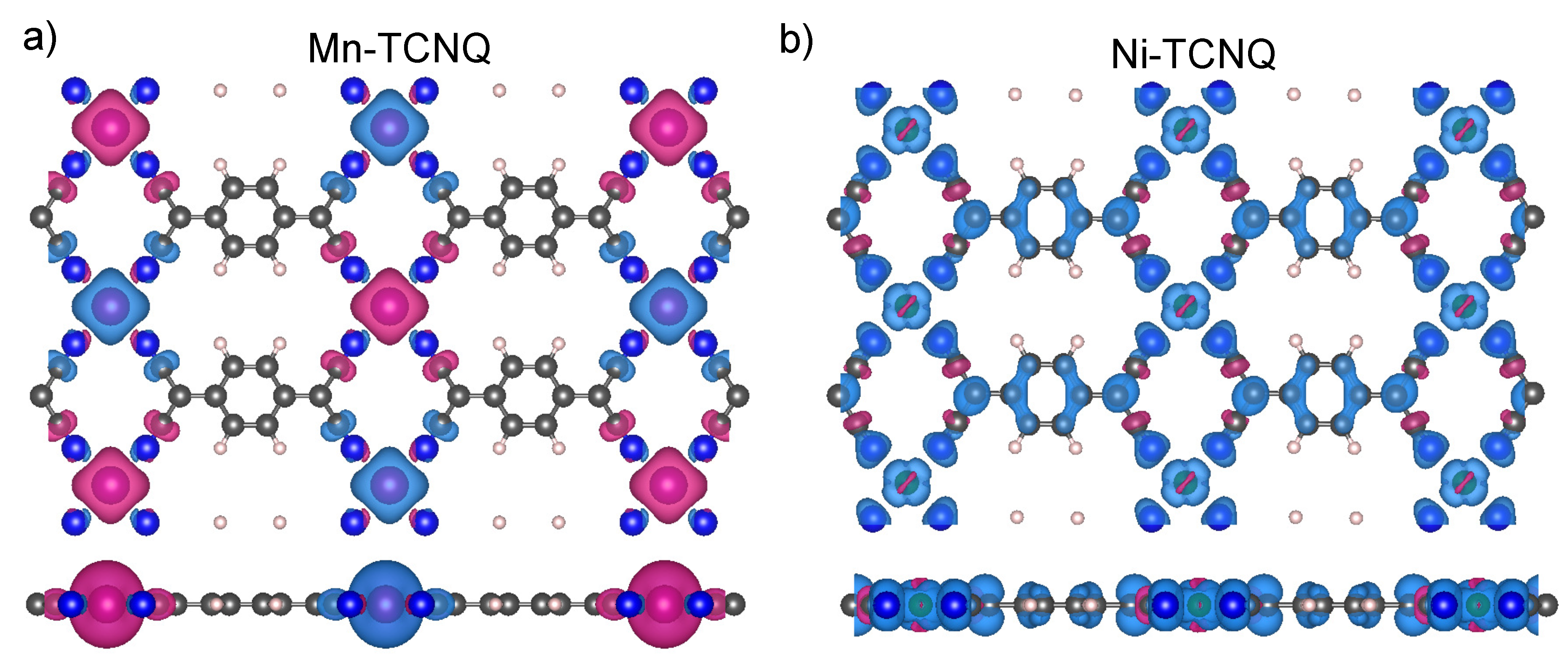
11]. The corresponding spin densities are shown in
Figure 8 for Mn–TCNQ and Ni–TCNQ. In
Section III of the Supplementary Material we also include other configurations obtained by using a rectangular
surface unit cell, in which other AFM configurations with spins aligned in rows or columns are considered as well [
32], showing the importance of next to nearest neighbors (diagonal) couplings in the networks that have been discussed in the previous section. We have obtained values of
J using the total energy differences between these frozen spin configurations (see
Supplementary Material Section III). The so-calculated values differ with respect to the fitted ones by a factor of five in the case of Mn–TCNQ and by two orders of magnitude in the case of Ni–TCNQ. The large discrepancy found in this latter case of Ni–TCNQ points again towards a more complex scenario than in the Mn–TCNQ case.
It is worth mentioning that the exchange constant
J obtained in [
11] refers to the coupling between the Ni spin and the itinerant spin density of the TCNQ LUMO hybridized band. That exchange coupling is of the direct exchange type and has, therefore, much larger typical values than the mediated couplings (RKKY and superexchange). To emphasize its direct exchange origin we denote it here by
. A rough estimate for the relation between the two exchange coupling constants can be obtained in terms of the band width of the LUMO hybrid band (
W) as
. Taking W ∼ 100 meV and the value
meV of [
11], we get
0.3 meV, which has the same order of magnitude as the fitted value.
3.4. Magnetocrystalline Anisotropy
The magnetocrystalline anisotropy energies (MAEs) can be obtained from DFT calculations that include SOC effects. The resulting total energies, thus, depend on the orientation of the magnetization density. For extended systems, where the transition metal atomic orbital momentum is expected to be partially or totally quenched, the MAE appears as a second-order SOC effect. In systems where the PDOS is characterized by sharp peaks and devoid of degeneracies at the Fermi level, a second-order perturbative treatment of the SOC makes it possible to establish a few guidelines for the likelihood of an easy axis or plane. The perturbation couples states above and below the Fermi level and it is inversely proportional to the energy difference between states. When the spin-up d-band is completely filled, it can be shown that the energy correction is proportional to the expected value of the orbital magnetic moment and that the spin–flip excitations are negligible [
33,
34,
35].
The total energy variation as a function of the magnetization axis direction is very subtle, often in the sub-meV range per atom. When spin–orbit effects are not strong, it is common practice to use the so-called second variational method [
36], where SOC is not treated self-consistently. First, a charge density is converged in a collinear spin-polarized calculation. Next, a new Hamiltonian that includes a SOC term is constructed and diagonalized for two different magnetization directions. Then, the MAE is calculated from the difference of the two band energies. Alternatively, a more precise MAE can be obtained from total energy calculations that include SOC self-consistently. Using the latter method, in this work we have calculated MAE values for free-standing Mn–TCNQ and Ni–TCNQ networks.
The small energies involved in the anisotropy are a challenge for DFT calculations. The MAE is highly sensitive to the geometry and electronic structure calculation details, such as the exchange and correlation functionals and basis set types. From a technical perspective, a reliable MAE is only achieved with demanding convergence criteria. For example, it has been observed that fine k-point samplings of the Brillouin zone are needed [
37,
38,
39]. An account of the convergence details as well as MAE dependence on the
U parameter can be found in
Section IV of the Supplementary Material.
Table 2 shows the obtained values for
eV in
cells (i.e., only ferromagnetic ordering is considered in this section). For the Mn–TCNQ rectangular network, we find in-plane magnetization with negligible azimuthal dependence, i.e., easy plane anisotropy. The MAE, calculated as the total energy difference between magnetic configurations with Mn magnetizations parallel to the OX and OZ axes, is 0.2 meV. In the Ni–TCNQ rectangular network the energetically preferred magnetization is out-of-plane and the MAE values vary significantly with the azimuthal direction. As shown in
Table 2, the values change as much as 1.50 meV with azimuthal angle variations.
The different behavior of the MAE with the azimuthal angle in Mn and Ni networks can be understood in terms of the differences in the metal–molecule bonds, particularly the Mn–N and Ni–N bonds. In both networks the d
(with magnetic quantum number
), d
, and d
orbitals remain rather localized, whereas the d
and d
orbitals are spread over a wider energy range of a few eV below the Fermi level (see
Figure 6 and
Figure 7). The delocalization of electronic charge in these d
and d
orbitals is stronger in the Ni–TCNQ case, where the latter two sub-bands are partially occupied and form hybrid states at the Fermi level with the TCNQ LUMO. As these hybrid states lie at the Fermi level, they have a dominant role in the magnetic anisotropy and, since they yield markedly directional charge and spin density distributions along the Ni–N bonds, they are likely to produce azimuthal MAE variations. Conversely, the Mn d-electrons hybridize weakly with the TCNQ orbitals close to the Fermi level, i.e., with the LUMO, and have essentially no weight at the Fermi level. The spatial extent of these relevant Ni–TCNQ hybrid states is manifested in the delocalized electron spin densities depicted in
Figure 8b, as compared to the case of Mn–TCNQ shown in
Figure 8a with a spin density more localized at the Mn sites and its neighboring cyano groups.
The existence of an easy axis (plane) of magnetization for Ni (Mn) cannot be anticipated from the electronic structure details. In the Mn–TCNQ system, since the d-band is half filled, the MAE is led by spin–flip excitations and, therefore, the value of the exchange splitting is determinant. In the absence of same-spin excitations, the anisotropy would be associated to the anisotropic part of the spin distribution. More precisely, the MAE would be proportional to the anisotropy of the expected values of the magnetic dipole operator [
34,
35]. However,
Figure 8a shows an anisotropic spin distribution extended towards the cyano groups of the organic ligand TCNQ in the network plane by the crystal field. The quadrupolar moment of this distribution should promote out-of-plane magnetization. This interpretation is at variance with the SOC-self-consistent DFT result. A more elaborated model has been proposed for systems with localized d-orbitals. It states that the spin–flip excitations that keep the quantum number
constant favor an in-plane magnetization [
40]. The calculated PDOS of
Figure 6 shows that the two
peaks (d
− d
) are those closer to the Fermi level, for majority and minority spin states, respectively. This situation is, in principle, compatible with an easy plane behavior. The conclusion we draw is that the basic qualitative feature of the magnetic anisotropy, namely the magnetization direction, cannot be accounted for by rules of general character, not even in a case like Mn–TCNQ, where the d-electrons have a rather localized character that would make this system seem a priori a good playground for these models.
Next, we turn our attention back to the case of Ni–TCNQ, where the DFT calculations yield a relatively large value for the MAE with out-of-plane magnetization, as well as significant variations of the MAE in the network plane. This theoretical result contrasts with the experimental absence of anisotropy in this system and, thus, requires a further analysis oriented at finding an explanation. As we discuss below, the discrepancy could be explained by substrate effects, mostly due to electronic charge transfer from the metal Au(111) surface. However, if we tried to calculate MAE values from DFT calculations with SOC using the supported Ni–TCNQ/Au(111) model structures presented above, we would not obtain informative results, since it would be very difficult in practice to disentangle the anisotropy effects originated by different aspects of the system. The most significant of them is the unavoidable artificial strain introduced in the system by forcing a commensurable Ni–TCNQ overlayer on top of the Au(111) surface due to the use of periodic boundary conditions in a finite size system imposed by our DFT calculations. However, these limitations can be more conveniently understood using free-standing models.
In the
rectangular Ni–TCNQ free-standing overlayer we can attribute the large MAE values to the partially occupied Ni(d
,d
) states. If these
bands were completely filled by transfer of 0.5 electrons from the metallic substrate, their contribution to the MAE would be dramatically reduced. Additionally, the Ni atom spin would become close to
, a case for which no single-ion anisotropy is possible [
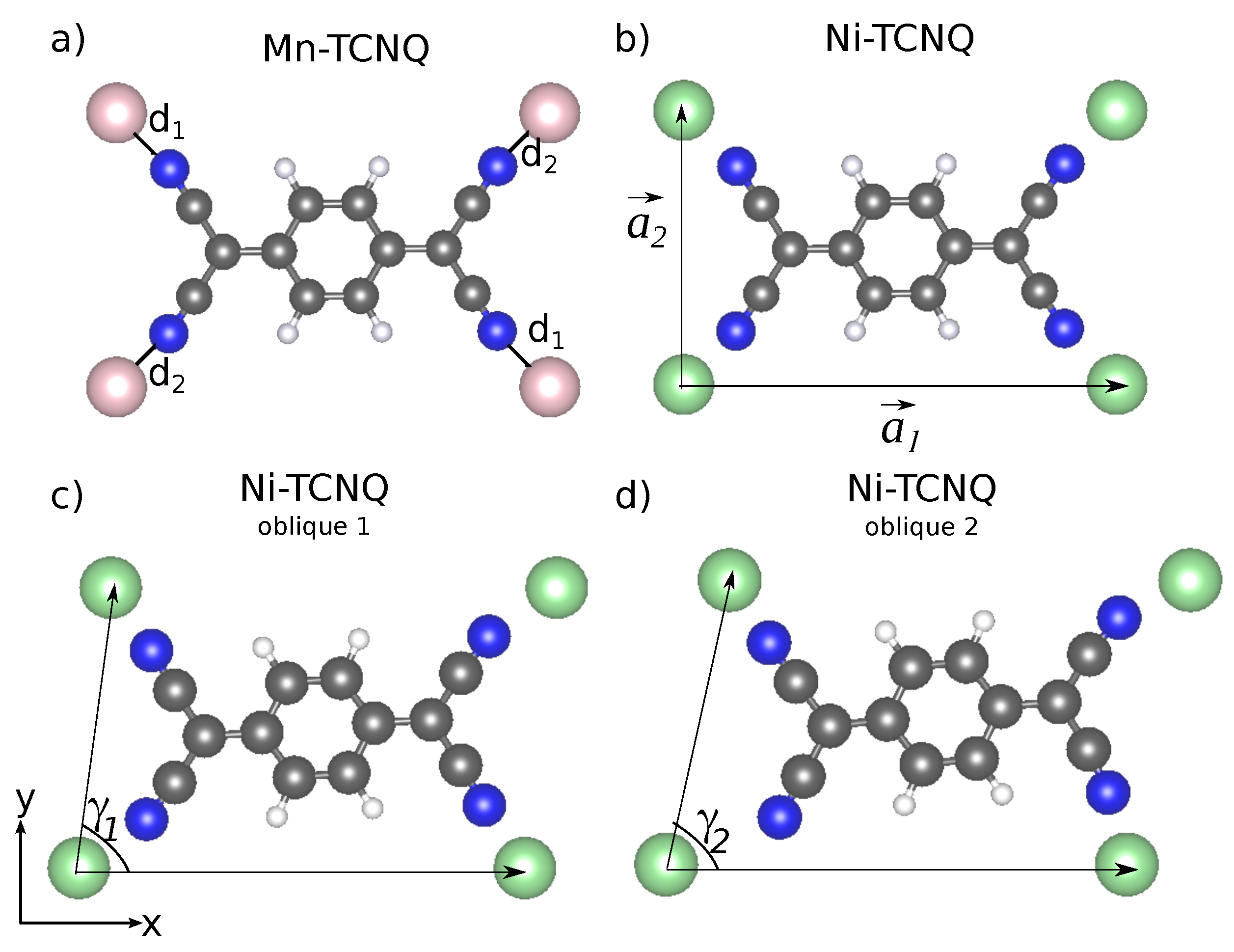
29]. However, it is hard to give a precise estimate of the amount of charge transfer and, on top of this, other sources of anisotropy reduction could be at play, like a reduction of Ni coordination due to a geometrical distortion. Indeed, the lowest-energy configuration of this rectangular unit cell is obtained upon a small symmetry-lowering distortion where the four Ni–N bonds are inequivalent: the bonds at
degrees with the
-axis (d
) have a length of 1.95 Å and the other pair at
(d
) of 2.01 Å (see
Figure 2). The former direction is that of the hardest magnetization axis. This symmetry breaking, though subtle from the geometry point of view, is nevertheless associated to a noticeable asymmetry in the electronic structure, which is in turn behind the strong azimuthal variability of the MAE. In a closer inspection of the PDOS we find that the Ni(d
,d
) peaks at the Fermi level hybridized with the molecule LUMO are contributed by d-orbitals lying on the plane containing the short Ni–N bonds (d
) and the surface normal (see
Section IV of the Supplementary Material). The long bonds (d
), to which
states at the Fermi level do not contribute, correspond to a softer magnetization direction.
To understand the consequences of this distorted geometry on the magnetic anisotropy, we have constructed a free-standing flat Ni–TCNQ model in an oblique unit cell, in which the angle
between the lattice vectors
and
is varied (the rectangular cell corresponds to
). As described in
Section IV of the Supplementary Material, two cases have been considered: a weakly distorted case with
and a larger distortion with
. The unit cell angle
has been reduced while uniformly scaling the lattice constants to keep the unit cell area equal to that of the rectangular equilibrium unit cell. Then, the atomic
coordinates have been relaxed to satisfy the same convergence criteria as in other models of the present work. For a larger distortion of the rectangular cell with
, one could force a commensurate supercell
on Au(111) [
4]. In the optimized structure the TCNQ is barely deformed, but one Ni–N bond at the azimuthal direction
is broken because of the cell distortion and the pair of bonds at the
direction have their lengths reduced to 1.85 Å (see
Section IV of the Supplementary Material). The magnetic anisotropy is significantly reduced with respect to that of the rectangular cell, but the hardest direction is still the one along the shortest pair or Ni–N bonds (see
Table 2). The main consequence of the Ni coordination reduction caused by the cell shape change is to partially quench its spin. We observe that the local magnetic moment is reduced by about 0.3
, approaching the ideal
state that would yield no anisotropy in the single-atom picture. We observe, nevertheless, that this distorted configuration still has partially filled Ni d
states at the Fermi level (see
Section IV of the Supplementary Material). Therefore, we note that this mechanism of anisotropy reduction and the charge transfer effect proposed above are of a different nature, although both originate from the interaction with the substrate.
All in all, the observed lack of magnetic anisotropy in the Ni-TCNQ/Au(111) XMCD data is clearly a substrate effect, which reduces the Ni–TCNQ anisotropy by a combined effect of charge transfer and change of coordination. Nonetheless, other subtle substrate effects not considered here might also have a role, such as fluctuations in the Au–Ni charge transfer due to the incommensurability and corrugation of the network.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}