
Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site


Abstract
:1. Introduction
1.1. Cancer
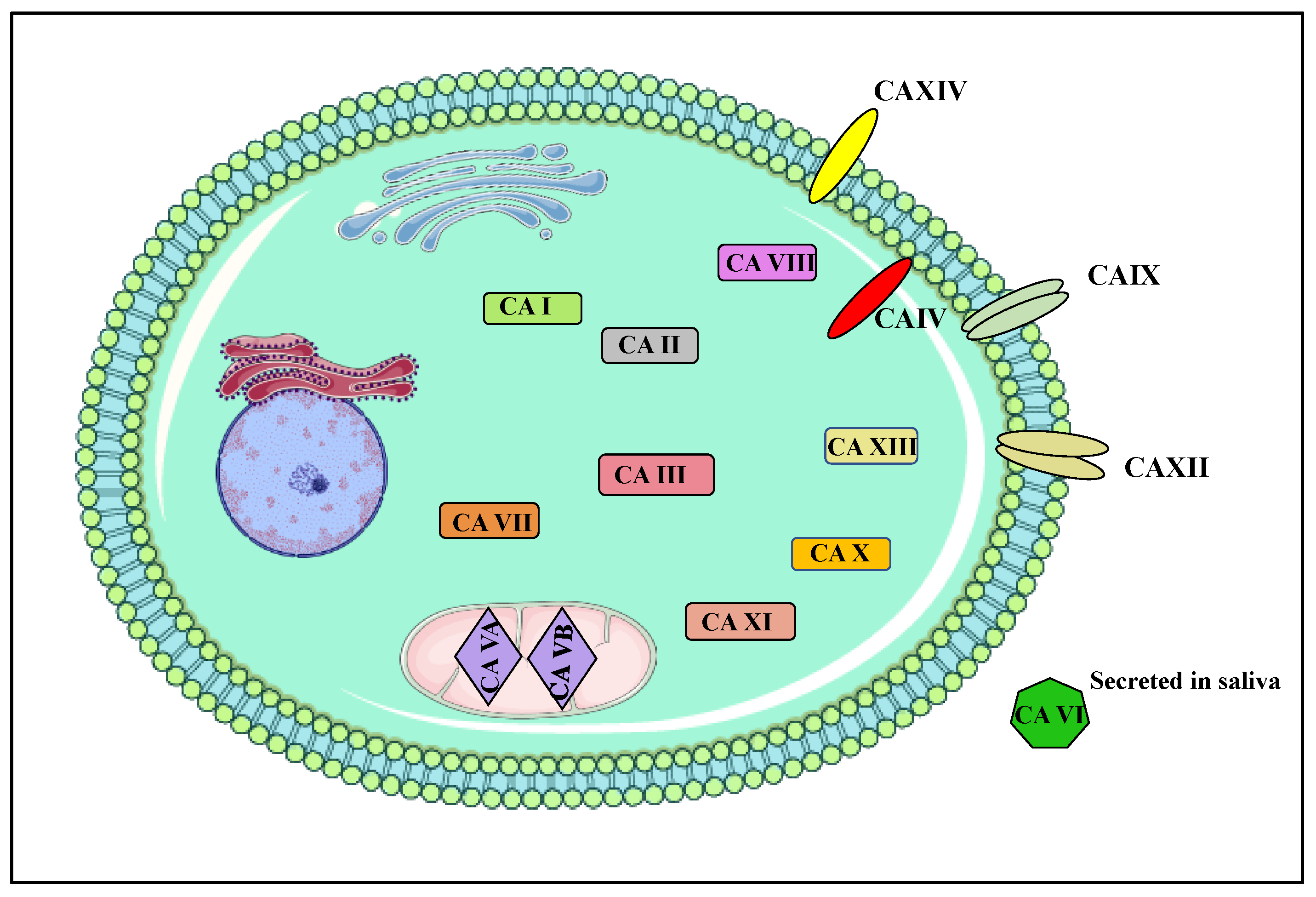
1.2. Carbonic Anhydrases
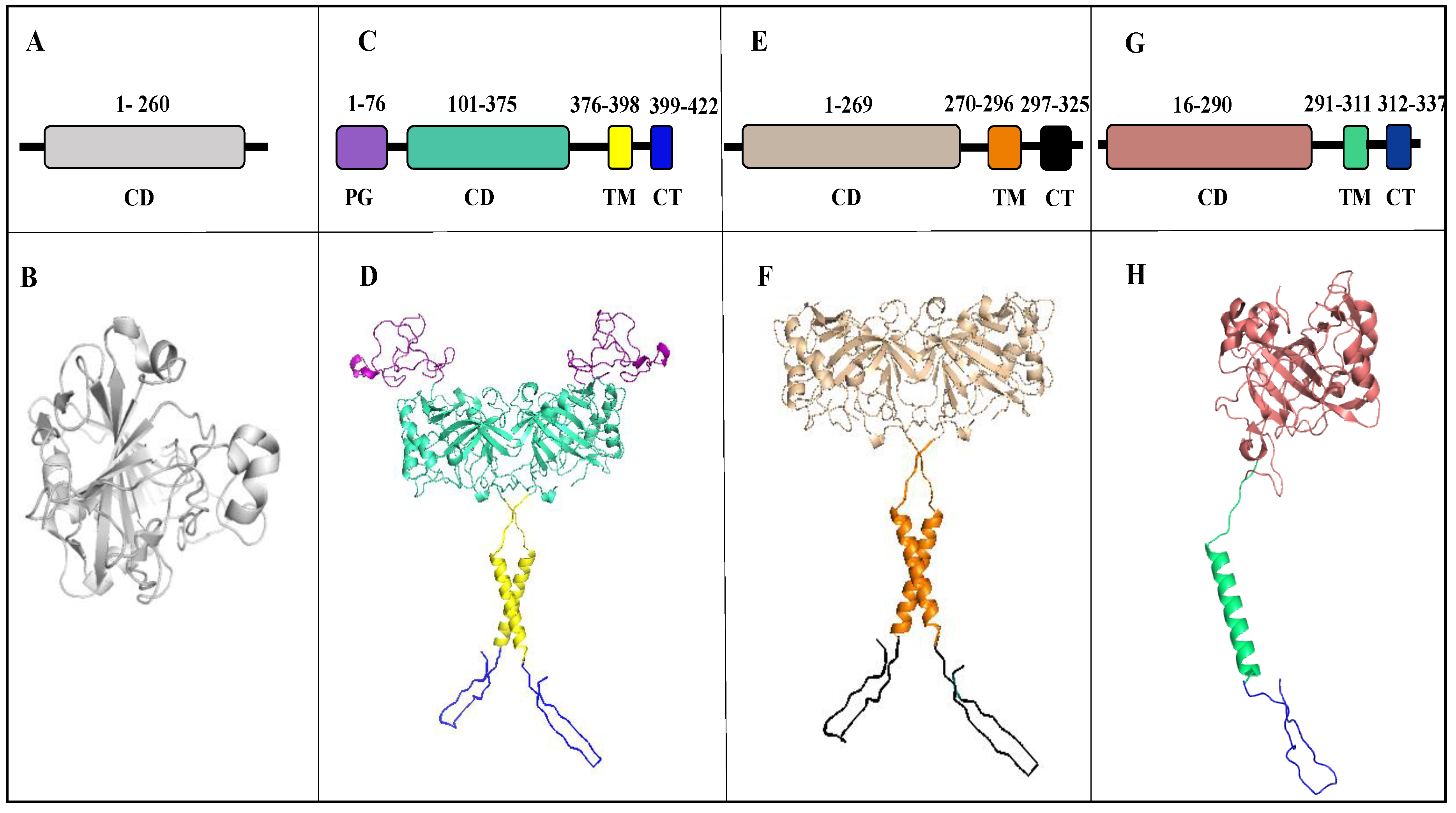
2. Structure
2.1. Carbonic Anhydrase IX
2.2. Carbonic Anhydrase XII
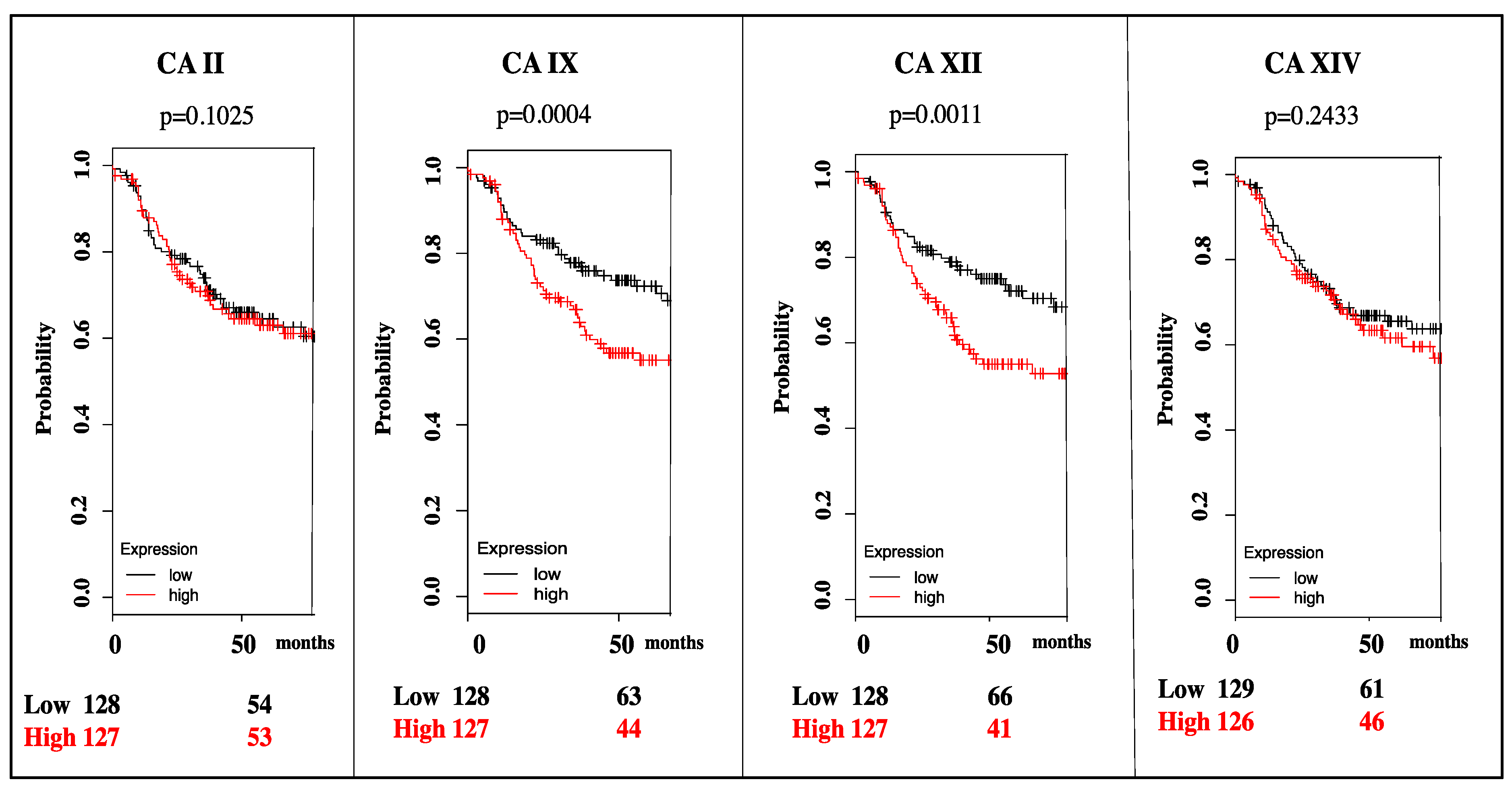
3. Targeting CA IX and CA XII Activity in Cancer
4. Mechanisms of CA Inhibition
4.1. Classical CAIs
4.2. Non-Classical CAIs
5. Isoform Specific Targeting of CAs in Cancer
5.1. Small Molecules in Clinical Trials
5.2. CA IX-Specific Monoclonal Antibodies for Immunotherapy and Immunodectection in Clinical Trials
5.3. CA XII-Specific Monoclonal Antibody
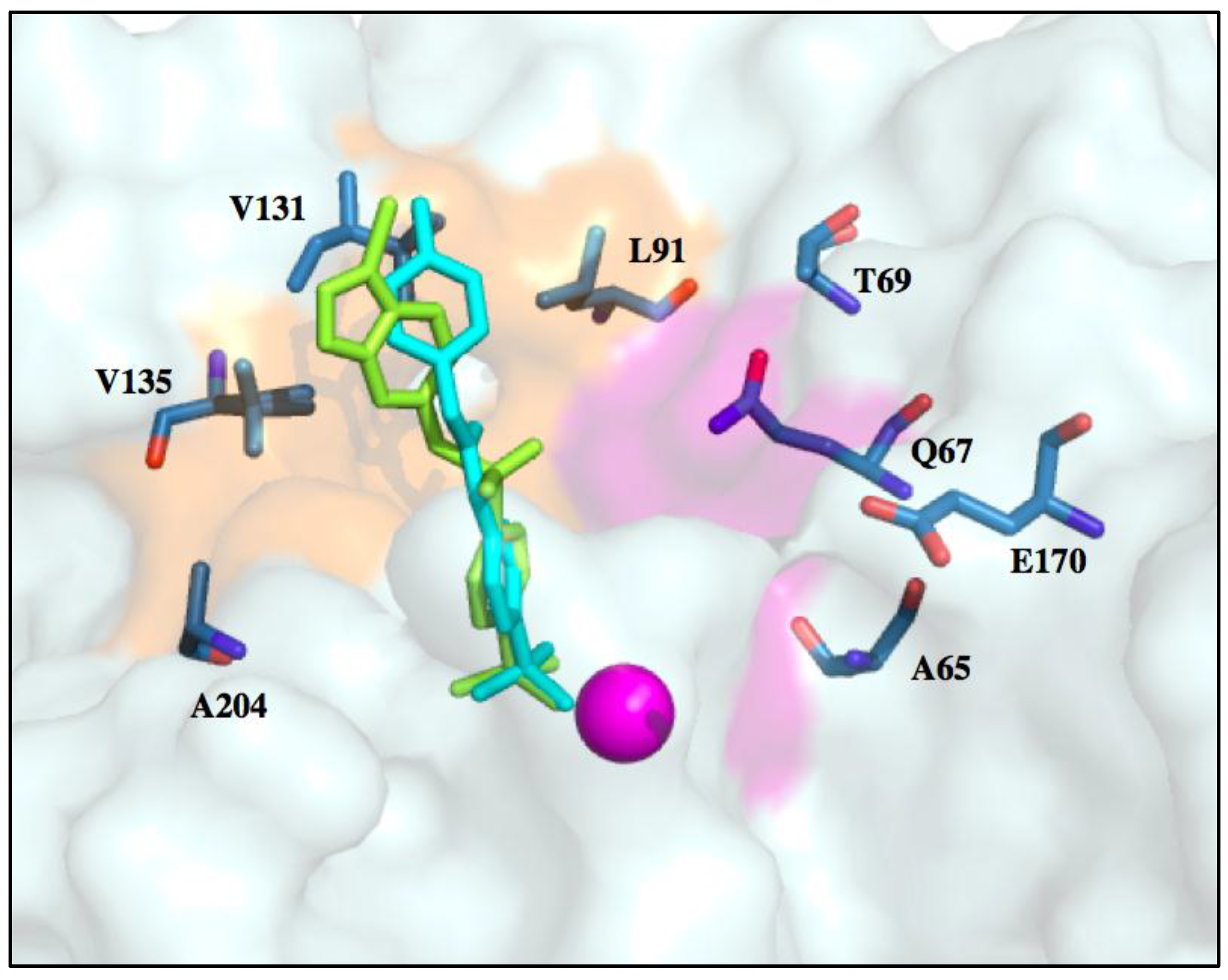
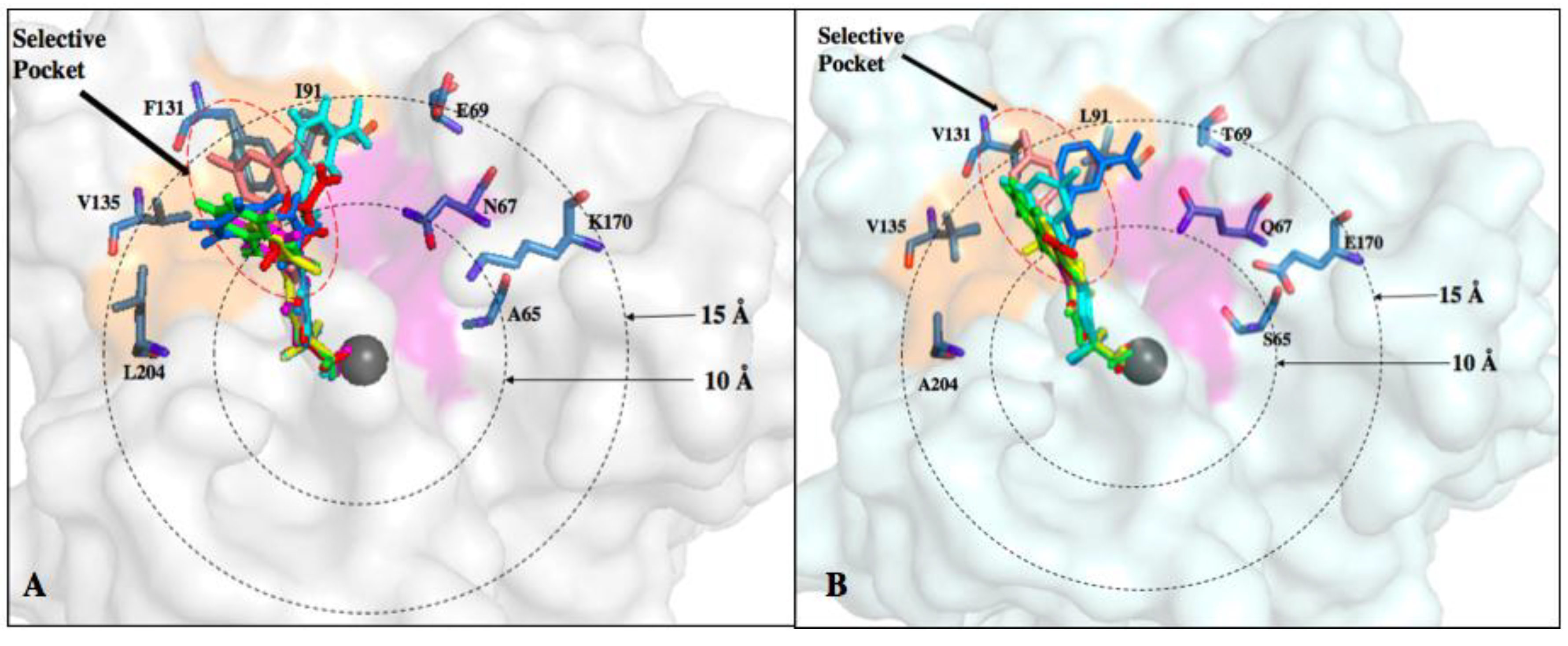
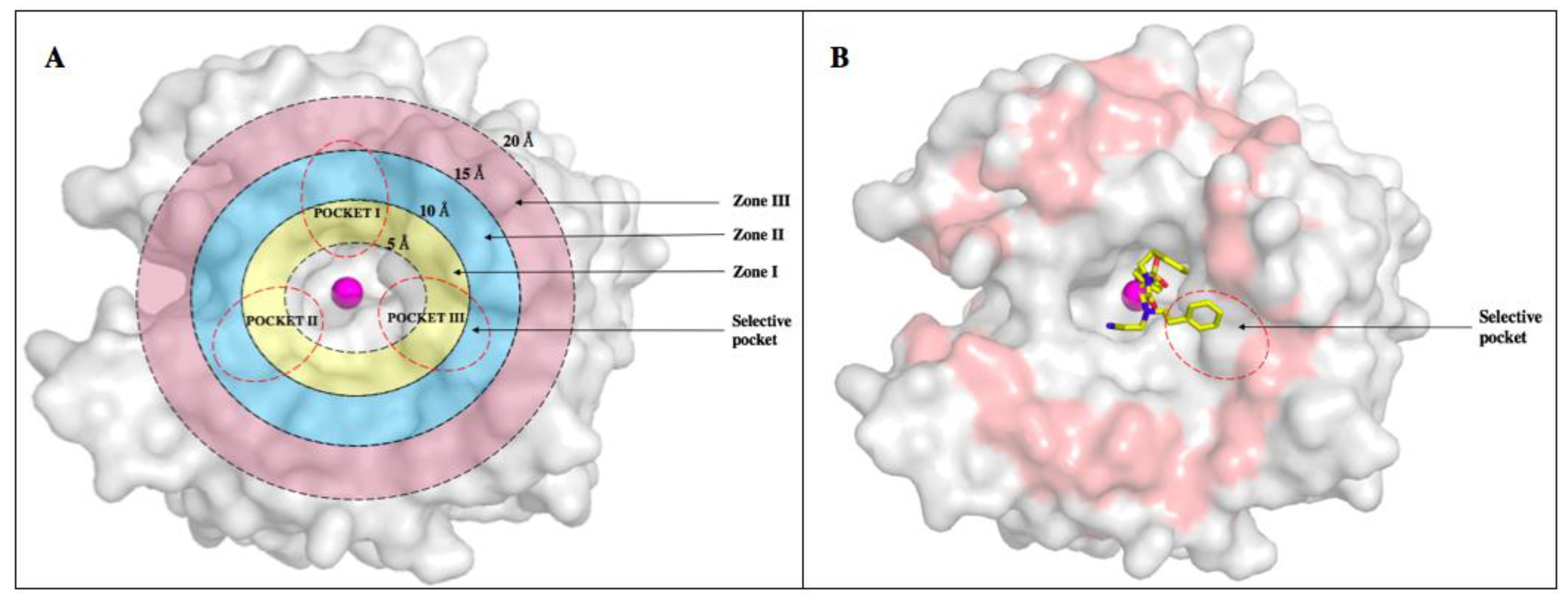
6. Improvements in Isoform Targeting of CA IX and CA XII
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- McDonald, P.C.; Winum, J.-Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting Carbonic Anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; McKenna, R.; Frost, S.C. Advances in anti-cancer drug development targeting Carbonic Anhydrase IX and XII. Top. Anti-Cancer Res. 2016, 5, 3–42. [Google Scholar]
- Pastorekova, S.; Supuran, C.T. Carbonic Anhydrase IX: From biology to therapy. In Hypoxia and Cancer; Cancer Drug Discovery and Development; Springer: New York, NY, USA, 2014; pp. 121–153. ISBN 978-1-4614-9166-8. [Google Scholar]
- Mahon, B.P.; Hendon, A.M.; Driscoll, J.M.; Rankin, G.M.; Poulsen, S.-A.; Supuran, C.T.; McKenna, R. Saccharin: A lead compound for structure-based drug design of carbonic anhydrase IX inhibitors. Bioorg. Med. Chem. 2015, 23, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Pinard, M.A.; McKenna, R. Targeting Carbonic Anhydrase IX activity and expression. Molecules 2015, 20, 2323–2348. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible Carbonic Anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Mahon, B.P.; Bhatt, A.; Socorro, L.; Driscoll, J.M.; Okoh, C.; Lomelino, C.L.; Mboge, M.Y.; Kurian, J.J.; Tu, C.; Agbandje-McKenna, M.; et al. The structure of Carbonic Anhydrase IX is adapted for low-pH catalysis. Biochemistry 2016, 55, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Zatovicova, M.; Pastorekova, S. Cancer-associated Carbonic Anhydrases and their inhibition. Curr. Pharm. Des. 2008, 14, 685–698. [Google Scholar] [CrossRef]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic anhydrase IX: Regulation and role in cancer. Subcell. Biochem. 2014, 75, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; Mahon, B.P.; Lamas, N.; Socorro, L.; Carta, F.; Supuran, C.T.; Frost, S.C.; McKenna, R. Structure activity study of Carbonic Anhydrase IX: Selective inhibition with ureido-substituted benzenesulfonamides. Eur. J. Med. Chem. 2017, 132, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Kallio, H.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Mannisto, S.; Heikinheimo, M.; Parkkila, S. Expression of Carbonic Anhydrases IX and XII during mouse embryonic development. BMC Dev. Biol. 2006, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Tafreshi, N.K.; Lloyd, M.C.; Bui, M.M.; Gillies, R.J.; Morse, D.L. Chapter 13: Carbonic Anhydrase IX as an imaging and therapeutic target for tumors and metastases. Subcell. Biochem. 2014, 75, 221–254. [Google Scholar] [CrossRef] [PubMed]
- Thiry, A.; Dogné, J.-M.; Masereel, B.; Supuran, C.T. Targeting tumor-associated Carbonic Anhydrase IX in cancer therapy. Trends Pharmacol. Sci. 2006, 27, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Sly, W.S. Carbonic Anhydrase XII functions in health and disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.; McKenna, R. Carbonic Anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Kallio, H. Regulation and Roles of Carbonic Anhydrases IX and XII; Tampere University Press: Tampere, Finland, 2011; ISBN 978-951-44-8622-7. [Google Scholar]
- Carbonic Anhydrase|Its Inhibitors and Activators|Taylor & Francis Group. Available online: https://www.taylorfrancis.com/books/e/9780203475300 (accessed on 11 January 2018).
- Hilvo, M.; Baranauskiene, L.; Salzano, A.M.; Scaloni, A.; Matulis, D.; Innocenti, A.; Scozzafava, A.; Monti, S.M.; Fiore, A.D.; Simone, G.D.; et al. Biochemical Characterization of CA IX, One of the Most Active Carbonic Anhydrase Isozymes. J. Biol. Chem. 2008, 283, 27799–27809. [Google Scholar] [CrossRef] [PubMed]
- Ondriskova, E.; Debreova, M.; Pastorekova, S. Chapter 10—Tumor-associated Carbonic Anhydrases IX and XII. In Carbonic Anhydrases as Biocatalysts; Supuran, C.T., Simone, G.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 169–205. ISBN 978-0-444-63258-6. [Google Scholar]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic Anhydrase IX: Regulation and role in cancer. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2014; pp. 199–219. ISBN 978-94-007-7358-5. [Google Scholar]
- Závada, J.; Závadová, Z.; Zat’ovičová, M.; Hyršl, L.; Kawaciuk, I. Soluble form of carbonic anhydrase IX (CA IX) in the serum and urine of renal carcinoma patients. Br. J. Cancer 2003, 89, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- İlie, M.; Mazure, N.M.; Hofman, V.; Ammadi, R.E.; Ortholan, C.; Bonnetaud, C.; Havet, K.; Venissac, N.; Mograbi, B.; Mouroux, J.; et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br. J. Cancer 2010, 102, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metast. Rev. 2007, 26, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Pastoreková, S.; Parkkila, S.; Parkkila, A.K.; Opavský, R.; Zelník, V.; Saarnio, J.; Pastorek, J. Carbonic Anhydrase IX, MN/CA IX: analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 1997, 112, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Saarnio, J.; Parkkila, S.; Parkkila, A.-K.; Pastoreková, S.; Haukipuro, K.; Pastorek, J.; Juvonen, T.; Karttunen, T.J. Transmembrane Carbonic Anhydrase, MN/CA IX, is a potential biomarker for biliary tumours. J. Hepatol. 2001, 35, 643–649. [Google Scholar] [CrossRef]
- Saarnio, J.; Parkkila, S.; Parkkila, A.K.; Waheed, A.; Casey, M.C.; Zhou, X.Y.; Pastoreková, S.; Pastorek, J.; Karttunen, T.; Haukipuro, K.; et al. Immunohistochemistry of Carbonic Anhydrase isozyme IX (MN/CA IX) in human gut reveals polarized expression in the epithelial cells with the highest proliferative capacity. J. Histochem. Cytochem. 1998, 46, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; auf dem Keller, U.; Leung, S.; Huntsman, D.; Clarke, B.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel Carbonic Anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed]
- Svastová, E.; Hulíková, A.; Rafajová, M.; Zat’ovicová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated Carbonic Anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767. [Google Scholar] [CrossRef] [PubMed]
- Vullo, D.; Franchi, M.; Gallori, E.; Pastorek, J.; Scozzafava, A.; Pastorekova, S.; Supuran, C.T. Carbonic Anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, cancer-associated isozyme IX with anions. J. Enzyme Inhib. Med. Chem. 2003, 18, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Sadri, N.; Zhang, P.J. Hypoxia-inducible factors: Mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers 2013, 5, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.P.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated Carbonic Anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Sedlakova, O.; Svastova, E.; Takacova, M.; Kopacek, J.; Pastorek, J.; Pastorekova, S. Carbonic Anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front. Physiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human Carbonic Anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Kuzmin, I.; Wei, M.-H.; Pack, S.; Geil, L.; Johnson, B.E.; Stanbridge, E.J.; Lerman, M.I. Down-regulation of transmembrane Carbonic Anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc. Natl. Acad. Sci. USA 1998, 95, 12596–12601. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Parkkila, S.; Zavada, J. Tumor-associated Carbonic Anhydrases and their clinical significance. In Advances in Clinical Chemistry; Elsevier: New York, NY, USA, 2006; Volume 42, pp. 167–216. [Google Scholar]
- Kivelä, A.J.; Kivelä, J.; Saarnio, J.; Parkkila, S. Carbonic Anhydrases in normal gastrointestinal tract and gastrointestinal tumours. World J. Gastroenterol. WJG 2005, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Winum, J.-Y. Drug Design of Zinc-Enzyme Inhibitors: Functional, Structural, and Disease Applications; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 978-0-470-50815-2. [Google Scholar]
- Pinard, M.A.; Mahon, B.; McKenna, R. Probing the surface of human Carbonic Anhydrase for clues towards the design of isoform specific inhibitors. BioMed Res. Int. 2015, 2015, 453543. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, A.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivelä, J.; Parkkila, A.-K.; Waheed, A.; Sly, W.S.; Grubb, J.H.; Shah, G.; et al. Expression of a novel transmembrane Carbonic Anhydrase isozyme XII in normal human gut and colorectal tumors. Am. J. Pathol. 2000, 156, 577–584. [Google Scholar] [CrossRef]
- Watson, P.H.; Chia, S.K.; Wykoff, C.C.; Han, C.; Leek, R.D.; Sly, W.S.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Carbonic Anhydrase XII is a marker of good prognosis in invasive breast carcinoma. Br. J. Cancer 2003, 88, 1065. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.H.; Sheng, S.; Charn, T.H.; Waheed, A.; Sly, W.S.; Lin, C.-Y.; Liu, E.T.; Katzenellenbogen, B.S. Estrogen receptor regulation of Carbonic Anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008, 68, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.; Liao, S.-Y.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; et al. Expression of hypoxia-inducible cell-surface transmembrane Carbonic Anhydrases in human cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar] [CrossRef]
- Hynninen, P.; Vaskivuo, L.; Saarnio, J.; Haapasalo, H.; Kivelä, J.; Pastoreková, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Puistola, U.; et al. Expression of transmembrane Carbonic Anhydrases IX and XII in ovarian tumours. Histopathology 2006, 49, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Gustafsson, J.-A. Estrogen receptor action. Crit. Rev. Eukaryot. Gene Expr. 2002, 12. [Google Scholar] [CrossRef]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Aggarwal, M.; Mahon, B.P.; Tu, C.; McKenna, R. A sucrose-binding site provides a lead towards an isoform-specific inhibitor of the cancer-associated enzyme carbonic anhydrase IX. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Pastoreková, S.; Callebaut, I.; Mornon, J.P.; Zelník, V.; Opavský, R.; Zat’ovicová, M.; Liao, S.; Portetelle, D.; Stanbridge, E.J. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994, 9, 2877–2888. [Google Scholar] [PubMed]
- Potter, C.P.S.; Harris, A.L. Diagnostic, prognostic and therapeutic implications of Carbonic Anhydrases in cancer. Br. J. Cancer 2003, 89, 2. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 978-0-470-12371-3. [Google Scholar]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Making a fast reaction faster: Carbonic Anhydrases. In Biochemistry; W H Freeman: New York, NY, USA, 2002; Volume 5. [Google Scholar]
- Xue, Y.; Liljas, A.; Jonsson, B.-H.; Lindskog, S. Structural analysis of the zinc hydroxide–Thr-199–Glu-106 hydrogen-bond network in human Carbonic Anhydrase II. Proteins Struct. Funct. Bioinform. 1993, 17, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Genis, C.; Sippel, K.H.; Case, N.; Cao, W.; Avvaru, B.S.; Tartaglia, L.J.; Govindasamy, L.; Tu, C.; Agbandje-McKenna, M.; Silverman, D.N.; et al. Design of a Carbonic Anhydrase IX active-site mimic to screen inhibitors for possible anti-cancer properties. Biochemistry 2009, 48, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Fisher, Z.S.; Maupin, M.C.; Budayova-Spano, M.; Govindasamy, L.; Tu, C.; Agbandje-McKenna, M.; Silverman, D.N.; Voth, G.A.; McKenna, R. Atomic crystal and molecular dynamics simulation structures of human Carbonic Anhydrase II: Insights into the proton transfer mechanism. Biochemistry 2007, 46, 2930–2937. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to Carbonic Anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, A.; Pastorekova, S.; Pastorek, J.; Scozzafava, A.; De Simone, G.; Supuran, C.T. The proteoglycan region of the tumor-associated Carbonic Anhydrase isoform IX acts as anintrinsic buffer optimizing CO2 hydration at acidic pH values characteristic of solid tumors. Bioorg. Med. Chem. Lett. 2009, 19, 5825–5828. [Google Scholar] [CrossRef] [PubMed]
- Ulmasov, B.; Waheed, A.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Tu, C.; Silverman, D.N. Purification and kinetic analysis of recombinant CA XII, a membrane Carbonic Anhydrase overexpressed in certain cancers. Proc. Natl. Acad. Sci. USA 2000, 97, 14212–14217. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Mahon, B.P.; Cruzeiro, V.W.D.; Cornelio, B.; Laronze-Cochard, M.; Ceruso, M.; Sapi, J.; Rance, G.A.; Khlobystov, A.N.; Fontana, A.; et al. Structure-activity relationships of benzenesulfonamide-based inhibitors towards Carbonic Anhydrase isoform specificity. Chembiochem. Eur. J. Chem. Biol. 2017, 18, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A.; Casini, A. Carbonic Anhydrase inhibitors. Med. Res. Rev. 2003, 23, 146–189. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many Carbonic Anhydrase inhibition mechanisms exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Winum, J.-Y. Designing Carbonic Anhydrase inhibitors for the treatment of breast cancer. Expert Opin. Drug Discov. 2015, 10, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.-Y.; Supuran, C.T. Recent advances in the discovery of zinc-binding motifs for the development of Carbonic Anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2015, 30, 321–324. [Google Scholar] [CrossRef] [PubMed]
- McKenna, R.; Supuran, C.T. Carbonic Anhydrase inhibitors drug design. Subcell. Biochem. 2014, 75, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A. Carbonic Anhydrase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2000, 10, 575–600. [Google Scholar] [CrossRef]
- Karioti, A.; Carta, F.; Supuran, C.T. Phenols and Polyphenols as Carbonic Anhydrase Inhibitors. Molecules 2016, 21, 1649. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Temperini, C.; Innocenti, A.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Polyamines inhibit Carbonic Anhydrases by anchoring to the zinc-coordinated water molecule. J. Med. Chem. 2010, 53, 5511–5522. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, R.; Pala, N.; Lomelino, C.; Mahon, B.P.; McKenna, R.; Dallocchio, R.; Dessì, A.; Carcelli, M.; Rogolino, D.; Sanna, V.; et al. Exploring Heteroaryl-pyrazole Carboxylic Acids as Human Carbonic Anhydrase XII Inhibitors. ACS Med. Chem. Lett. 2017, 8, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-zinc mediated inhibition of Carbonic Anhydrases: Coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Tars, K.; Vullo, D.; Kazaks, A.; Leitans, J.; Lends, A.; Grandane, A.; Zalubovskis, R.; Scozzafava, A.; Supuran, C.T. Sulfocoumarins (1,2-Benzoxathiine-2,2-dioxides): A class of potent and isoform-selective inhibitors of tumor-associated Carbonic Anhydrases. J. Med. Chem. 2013, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; Supuran, C.T.; McKenna, R. Non-classical inhibition of Carbonic Anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Ludwig, P.A.; Christianson, D.W. Two-site binding of phenol in the active site of human Carbonic Anhydrase II: Structural implications for substrate association. J. Am. Chem. Soc. 1994, 116, 3659–3660. [Google Scholar] [CrossRef]
- Maresca, A.; Akyuz, G.; Osman, S.M.; AlOthman, Z.; Supuran, C.T. Inhibition of mammalian carbonic anhydrase isoforms I–XIV with a series of phenolic acid esters. Bioorg. Med. Chem. 2015, 23, 7181–7188. [Google Scholar] [CrossRef] [PubMed]
- Langella, E.; D’Ambrosio, K.; D’Ascenzio, M.; Carradori, S.; Monti, S.M.; Supuran, C.T.; De Simone, G. A Combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of Carbonic Anhydrases. Chem. Eur. J. 2016, 22, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Cohen, S.M. Nucleophile recognition as an alternative inhibition mode for benzoic acid based Carbonic Anhydrase inhibitors. Chem. Commun. 2012, 48, 5259–5261. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, K.; Carradori, S.; Monti, S.M.; Buonanno, M.; Secci, D.; Vullo, D.; Supuran, C.T.; Simone, G.D. Out of the active site binding pocket for Carbonic Anhydrase inhibitors. Chem. Commun. 2014, 51, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.-M.; El-Azab, A.S.; Ceruso, M.; Supuran, C.T. Carbonic Anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds. Bioorg. Med. Chem. Lett. 2014, 24, 5185–5189. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of Carbonic Anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Touisni, N.; Maresca, A.; McDonald, P.C.; Lou, Y.; Scozzafava, A.; Dedhar, S.; Winum, J.-Y.; Supuran, C.T. Glycosyl coumarin Carbonic Anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J. Med. Chem. 2011, 54, 8271–8277. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Vullo, D.; Osman, S.M.; AlOthman, Z.; Supuran, C.T. Synthesis and Carbonic Anhydrase inhibition of a series of SLC-0111 analogs. Bioorg. Med. Chem. 2017, 25, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Haddad, R.I.; Weinstein, L.J.; Wieczorek, T.J.; Bhattacharya, N.; Raftopoulos, H.; Oster, M.W.; Zhang, X.; Latham, V.M.; Costello, R.; Faucher, J.; et al. A phase II clinical and pharmacodynamic study of E7070 in patients with metastatic, recurrent, or refractory squamous cell carcinoma of the head and neck: Modulation of retinoblastoma protein phosphorylation by a novel chloroindolyl sulfonamide cell cycle inhibitor. Clin. Cancer Res. 2004, 10, 4680–4687. [Google Scholar] [CrossRef] [PubMed]
- Abbate, F.; Casini, A.; Owa, T.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg. Med. Chem. Lett. 2004, 14, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.-Y.; Poulsen, S.-A.; Supuran, C.T.; Supuran, C.T. Therapeutic applications of the carbonic anhydrase inhibitors. Med. Res. Rev. 2009, 29, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Oosterwijk-Wakka, J.C.; Boerman, O.C.; Mulders, P.F.A.; Oosterwijk, E. Application of monoclonal antibody G250 recognizing Carbonic Anhydrase IX in renal cell carcinoma. Int. J. Mol. Sci. 2013, 14, 11402–11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, C.; Harris, A.L. Hypoxia inducible Carbonic Anhydrase IX, marker of tumour: Hypoxia, survival pathway and therapy target. Cell Cycle 2004, 3, 159–162. [Google Scholar] [CrossRef]
- Lau, J.; Liu, Z.; Lin, K.-S.; Pan, J.; Zhang, Z.; Vullo, D.; Supuran, C.T.; Perrin, D.M.; Bénard, F. Trimeric radiofluorinated sulfonamide derivatives to achieve in vivo selectivity for Carbonic Anhydrase IX-targeted PET imaging. J. Nucl. Med. 2015, 56, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.J.; Niemans, R.; van Kuijk, S.J.A.; Panth, K.M.; Parvathaneni, N.-K.; Peeters, S.G.J.A.; Zegers, C.M.L.; Rekers, N.H.; van Gisbergen, M.W.; Biemans, R.; et al. New ways to image and target tumour hypoxia and its molecular responses. Radiother. Oncol. 2015, 116, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Clinical Study to Evaluate the Maximum Tolerated Dose of BAY79-4620 Given Every 2 Weeks to Patients with Advanced Solid Tumors—Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT01065623 (accessed on 16 January 2018).
- Petrul, H.M.; Schatz, C.A.; Kopitz, C.C.; Adnane, L.; McCabe, T.J.; Trail, P.; Ha, S.; Chang, Y.S.; Voznesensky, A.; Ranges, G.; et al. Therapeutic mechanism and efficacy of the antibody–drug conjugate BAY 79-4620 targeting human Carbonic Anhydrase 9. Mol. Cancer Ther. 2012, 11, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.M.; Wu, A.M. Advances in immuno–positron emission tomography: Antibodies for molecular imaging in oncology. J. Clin. Oncol. 2012, 30, 3884–3892. [Google Scholar] [CrossRef] [PubMed]
- Divgi, C.R.; Pandit-Taskar, N.; Jungbluth, A.A.; Reuter, V.E.; Gönen, M.; Ruan, S.; Pierre, C.; Nagel, A.; Pryma, D.A.; Humm, J.; et al. Preoperative characterisation of clear-cell renal carcinoma using iodine-124-labelled antibody chimeric G250 (124I-cG250) and PET in patients with renal masses: A phase I trial. Lancet Oncol. 2007, 8, 304–310. [Google Scholar] [CrossRef]
- Povoski, S.P.; Hall, N.C.; Murrey, D.A.; Sharp, D.S.; Hitchcock, C.L.; Mojzisik, C.M.; Bahnson, E.E.; Knopp, M.V.; Martin, E.W.; Bahnson, R.R. Multimodal imaging and detection strategy with 124 I-labeled chimeric monoclonal antibody cG250 for accurate localization and confirmation of extent of disease during laparoscopic and open surgical resection of clear cell renal cell carcinoma. Surg. Innov. 2013, 20, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Oosterwijk, E.; Boerman, O.C.; Oyen, W.J.C.; Old, L.J.; Mulders, P.F.A. Antibody therapy in renal cell carcinoma. World J. Urol. 2008, 26, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winum, J.-Y.; Capasso, C. Novel antibody to a carbonic anhydrase: Patent evaluation of WO2011138279A1. Expert Opin. Ther. Pat. 2013, 23, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Dekaminaviciute, D.; Kairys, V.; Zilnyte, M.; Petrikaite, V.; Jogaite, V.; Matuliene, J.; Gudleviciene, Z.; Vullo, D.; Supuran, C.T.; Zvirbliene, A. Monoclonal antibodies raised against 167–180 aa sequence of human carbonic anhydrase XII inhibit its enzymatic activity. J. Enzyme Inhib. Med. Chem. 2014, 29, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Battke, C.; Kremmer, E.; Mysliwietz, J.; Gondi, G.; Dumitru, C.; Brandau, S.; Lang, S.; Vullo, D.; Supuran, C.; Zeidler, R. Generation and characterization of the first inhibitory antibody targeting tumour-associated carbonic anhydrase XII. Cancer Immunol. Immunother. CII 2011, 60, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gondi, G.; Mysliwietz, J.; Hulikova, A.; Jen, J.P.; Swietach, P.; Kremmer, E.; Zeidler, R. Antitumor efficacy of a monoclonal antibody that inhibits the activity of cancer-associated carbonic anhydrase XII. Cancer Res. 2013, 73, 6494–6503. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Barathova, M.; Kopacek, J.; Pastorek, J. Carbonic Anhydrase inhibitors targeting cancer: Therapeutic, immunologic, and diagnostic tools targeting isoforms IX and XII. In Drug Design of Zinc-Enzyme Inhibitors; Supuran, C.T., Winum, J.-Y., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 193–222. ISBN 978-0-470-50816-9. [Google Scholar]
- Casey, J.R.; Morgan, P.E.; Vullo, D.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J. Med. Chem. 2004, 47, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Groves, K.; Bao, B.; Zhang, J.; Handy, E.; Kennedy, P.; Cuneo, G.; Supuran, C.T.; Yared, W.; Peterson, J.D.; Rajopadhye, M. Synthesis and evaluation of near-infrared fluorescent sulfonamide derivatives for imaging of hypoxia-induced carbonic anhydrase IX expression in tumors. Bioorg. Med. Chem. Lett. 2012, 22, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Maren, T.H. Carbonic anhydrase: Chemistry, physiology, and inhibition. Physiol. Rev. 1967, 47, 595–781. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Paul, B.; Hofmann, A.; Morizzi, J.; Wu, Q.K.; Charman, S.A.; Nocentini, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. S-Glycosyl primary sulfonamides−A new structural class for selective inhibition of cancer-associated Carbonic Anhydrases. J. Med. Chem. 2009, 52, 6421–6432. [Google Scholar] [CrossRef] [PubMed]
- Carroux, C.J.; Rankin, G.M.; Moeker, J.; Bornaghi, L.F.; Katneni, K.; Morizzi, J.; Charman, S.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. A prodrug approach toward cancer-related Carbonic Anhydrase inhibition. J. Med. Chem. 2013, 56, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Supuran, C.T.; Poulsen, S.-A. A novel class of carbonic anhydrase inhibitors: Glycoconjugate benzene sulfonamides prepared by “click-tailing. ” J. Med. Chem. 2006, 49, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Vitavska, O.; Wieczorek, H. Identification of an animal sucrose transporter. J. Cell Sci. 2011, 124, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Trajkovic, J.; Bornaghi, L.F.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A. Design, synthesis, and biological evaluation of novel carbohydrate-based sulfamates as carbonic anhydrase inhibitors. J. Med. Chem. 2011, 54, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Eck, P.; Chen, S.; Corpe, C.P.; Lee, J.-H.; Kruhlak, M.; Levine, M. Inhibition of the intestinal glucose transporter GLUT2 by flavonoids. FASEB J. 2007, 21, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Köhler, K.; Hillebrecht, A.; Schulze Wischeler, J.; Innocenti, A.; Heine, A.; Supuran, C.T.; Klebe, G. Saccharin inhibits carbonic anhydrases: Possible explanation for its unpleasant metallic aftertaste. Angew. Chem. Int. Ed. 2007, 46, 7697–7699. [Google Scholar] [CrossRef] [PubMed]
- Morkūnaitė, V.; Baranauskienė, L.; Zubrienė, A.; Kairys, V.; Ivanova, J.; Trapencieris, P.; Matulis, D. Saccharin sulfonamides as inhibitors of Carbonic Anhydrases I, II, VII, XII, and XIII. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzio, M.; Guglielmi, P.; Carradori, S.; Secci, D.; Florio, R.; Mollica, A.; Ceruso, M.; Akdemir, A.; Sobolev, A.P.; Supuran, C.T. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J. Enzyme Inhib. Med. Chem. 2017, 32, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Tanc, M.; Ivanova, J.; Zalubovskis, R.; Vozny, I.; Monti, S.M.; Di Fiore, A.; De Simone, G.; Supuran, C.T. X-ray crystallographic and kinetic investigations of 6-sulfamoyl-saccharin as a carbonic anhydrase inhibitor. Org. Biomol. Chem. 2015, 13, 4064–4069. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int. J. Mol. Sci. 2013, 14, 24706–24725. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CA Inhibitors(CAI) | Structure | Ki (M) * | Reference | ||
|---|---|---|---|---|---|
| CA II | CA IX | CA XII | |||
| Classical CAI | |||||
| Sulfonamide (acetazolamide) |  | 0.012 | 0.025 | 0.0057 | [8] |
| Non-Classical CAI | |||||
| Phenolic (Phenol) |  | 5.5 | 8.8 | 9.2 | [75] |
| Polyamine (Spermine) |  | 84 | 13.3 | 27.6 | [76] |
| Carboxylic acid (3-(1-Ethyl-1H-indol-3-yl) -1H-pyrazole-5-carboxylic acid) |  | 1820 | 7.8 | 7.8 | [77] |
| Coumarins (6-(1S-Hydroxy-3 methylbutyl)-7-methoxy-2H-chromen-2-one) |  | 0.06 | 54.5 | [78] | |
| Sulfocoumarin (6-Hydroxy 1,2-Benzoxanthiine-2,2-dioxides) |  | >100 | 0.3 | 0.2 | [79] |
| Ki (nM) | |||
|---|---|---|---|
| Compounds | CA II | CA IX | CA XII |
| SLC-0111 | 960 | 45 | 5 |
| E7070 | 15 | 24 | 3 |
| Clinical Trail | CTID | Treatment | Status (2018) |
|---|---|---|---|
| Small Molecule | |||
| SLC-0111 | NCT02215850 | Advanced solid tumors | Phase I complete |
| E7070 | NCT00003891 | Solid tumors | Phase I complete |
| E7070 | NCT00080197 | Metastatic breast cancer | Phase II complete |
| E7070 | NTC0169197 | Relapsed AML and High-Risk Myelodysplastic Syndromes | Phase II complete |
| Monoclonal antibodies | |||
| Girentuximab(cG250) | NCT00087022 | Patients undergoing non-metatstatic kidney cancer | Phase III complete |
| BAY 79-4620 | NCT01028755 | Advance stage tumor | Phase I complete |
| I131-cG250 | NCT00003102 | Kidney cancer | Phase I complete |
| Imaging | |||
| Zr89-girentuximab PET/CT | NCT02883153 | Renal cell carcinoma | Phase III complete |
| In111-DOTA-girentuximab-IRDye800CW | NCT02497599 | Renal cell carcinoma | Recruiting |
| I 124-cG250 | NCT00606632 | Renal cell carcinoma | Phase III complete |
| Residue Number * | Distance from Zinc () | CA I | CA II | CA IX | CA XII |
|---|---|---|---|---|---|
| 5–10 Å | |||||
| 62 | 9.1 | Val | Asn | Asn | Asn |
| 65 | 6.9 | Ser | Ala | Ser | Ser |
| 67 | 7.3 | His | Asn | Gln | Lys |
| 10–15 Å | |||||
| 60 | 13.7 | Ile | Leu | Arg | Thr |
| 69 | 13.8 | Asn | Glu | Thr | Asn |
| 91 | 11.1 | Phe | Ile | Leu | Thr |
| 131 | 10.4 | Leu | Phe | Val | Ala |
| 135 | 12.2 | Ala | Val | Leu | Ser |
| 204 | 13.7 | Tyr | Leu | Ala | Asn |
| 15–20 Å | |||||
| 19 | 19.1 | Leu | Asp | Val | Lys |
| 20 | 15.2 | Tyr | Phe | Ser | Tyr |
| 57 | 19.6 | Lys | Leu | Leu | Phe |
| 58 | 16.3 | Glu | Arg | Arg | Leu |
| 71 | 21.1 | Glu | Asp | Pro | Pro |
| 72 | 15.9 | Asp | Asp | Pro | Ser |
| 123 | 15.4 | Trp | Trp | Leu | Tyr |
| 130 | 19.1 | Ser | Asp | Arg | Asp |
| 132 | 17.3 | Ala | Gly | Asp | Ser |
| 136 | 17.6 | Ser | Gln | Gly | Asn |
| 170 | 18.9 | Lys | Lys | Ser | Lys |
| 173 | 19.6 | Arg | Ser | Glu | Glu |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. https://doi.org/10.3390/molecules23051045
Singh S, Lomelino CL, Mboge MY, Frost SC, McKenna R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules. 2018; 23(5):1045. https://doi.org/10.3390/molecules23051045
Chicago/Turabian StyleSingh, Srishti, Carrie L. Lomelino, Mam Y. Mboge, Susan C. Frost, and Robert McKenna. 2018. "Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site" Molecules 23, no. 5: 1045. https://doi.org/10.3390/molecules23051045
APA StyleSingh, S., Lomelino, C. L., Mboge, M. Y., Frost, S. C., & McKenna, R. (2018). Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules, 23(5), 1045. https://doi.org/10.3390/molecules23051045