
Fingerprint-Based Machine Learning Approach to Identify Potent and Selective 5-HT2BR Ligands

 and
and 
Abstract
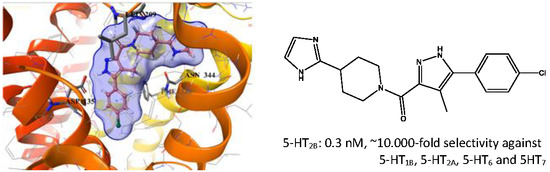
:
1. Introduction
2. Results and Discussion
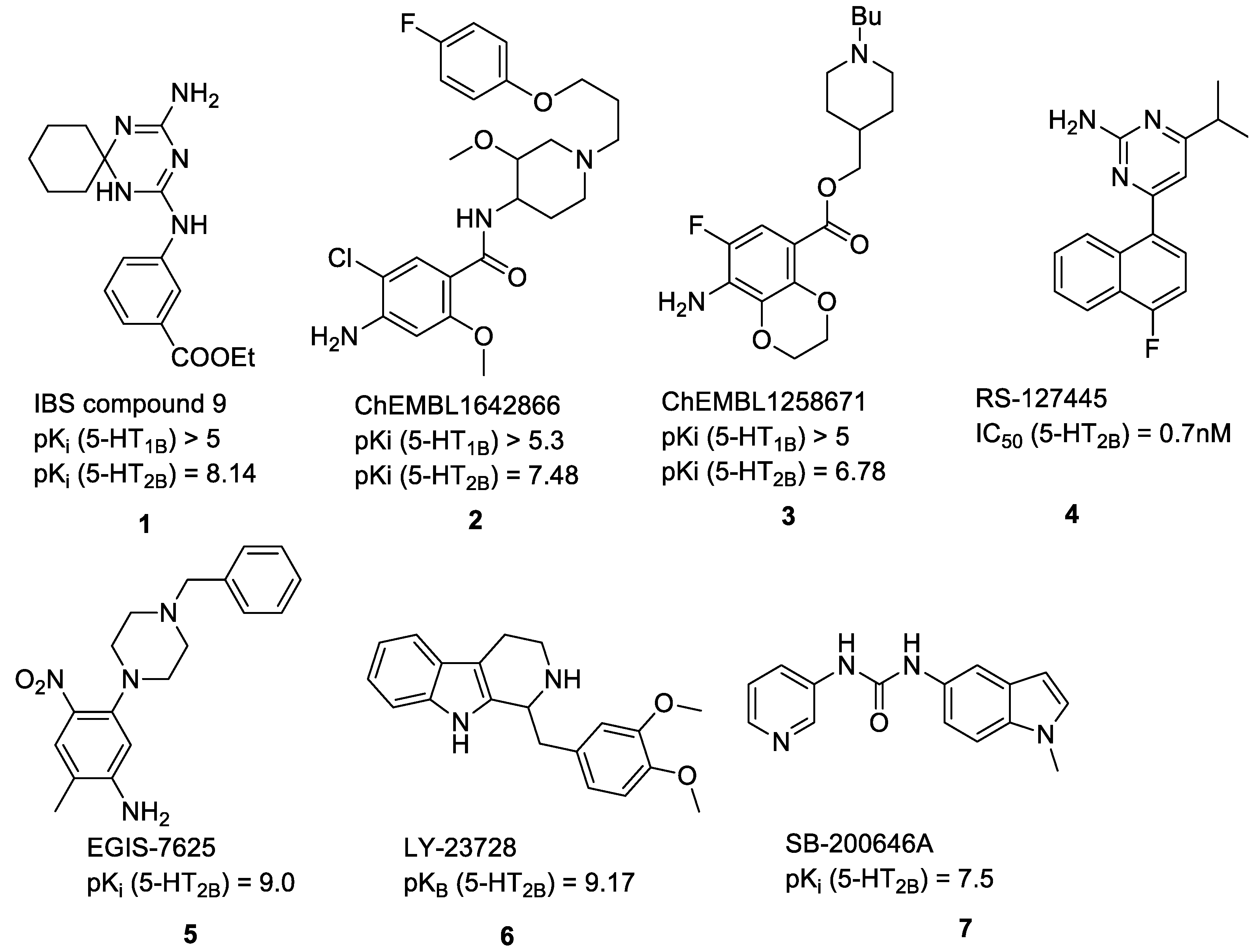
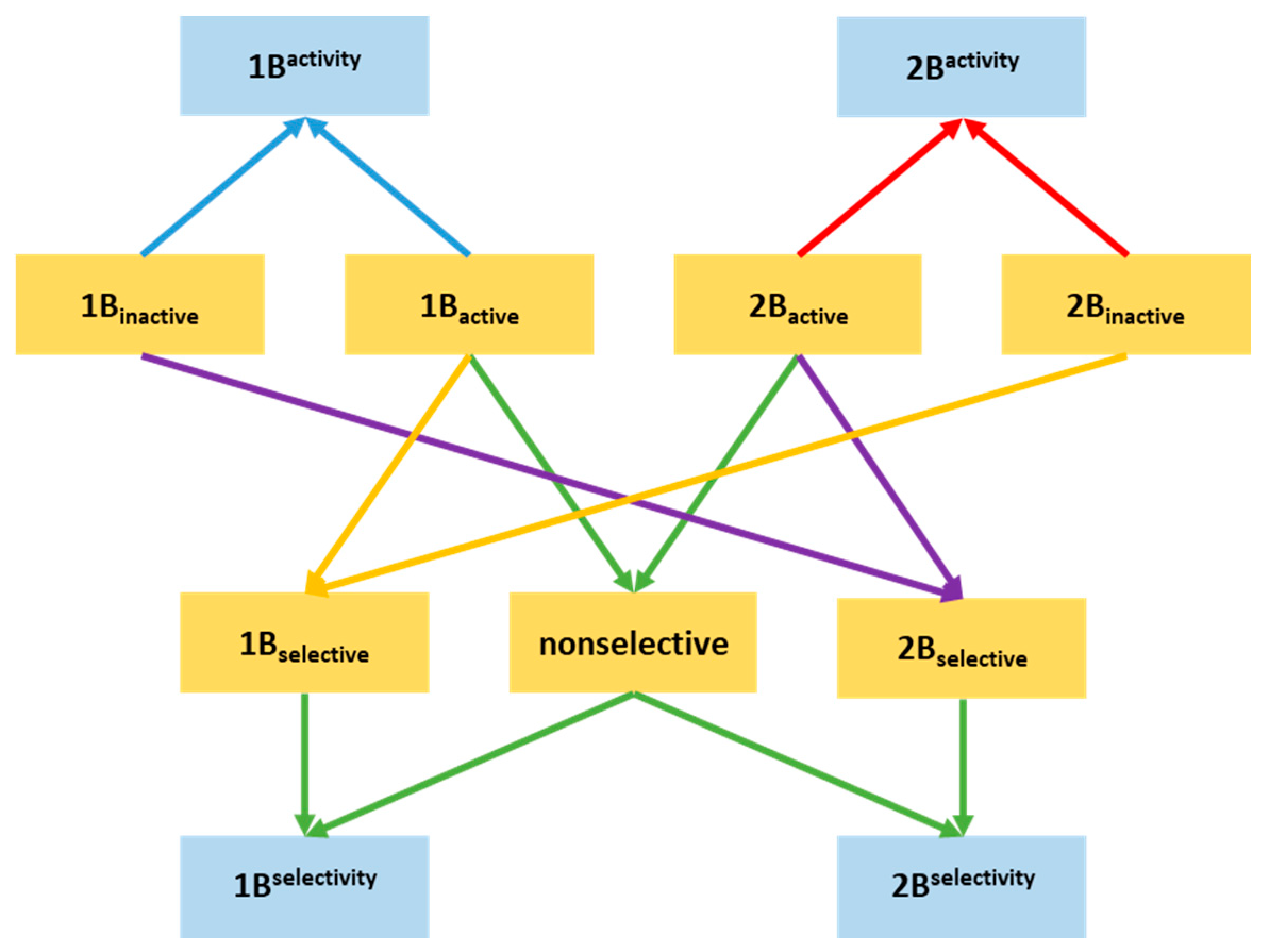
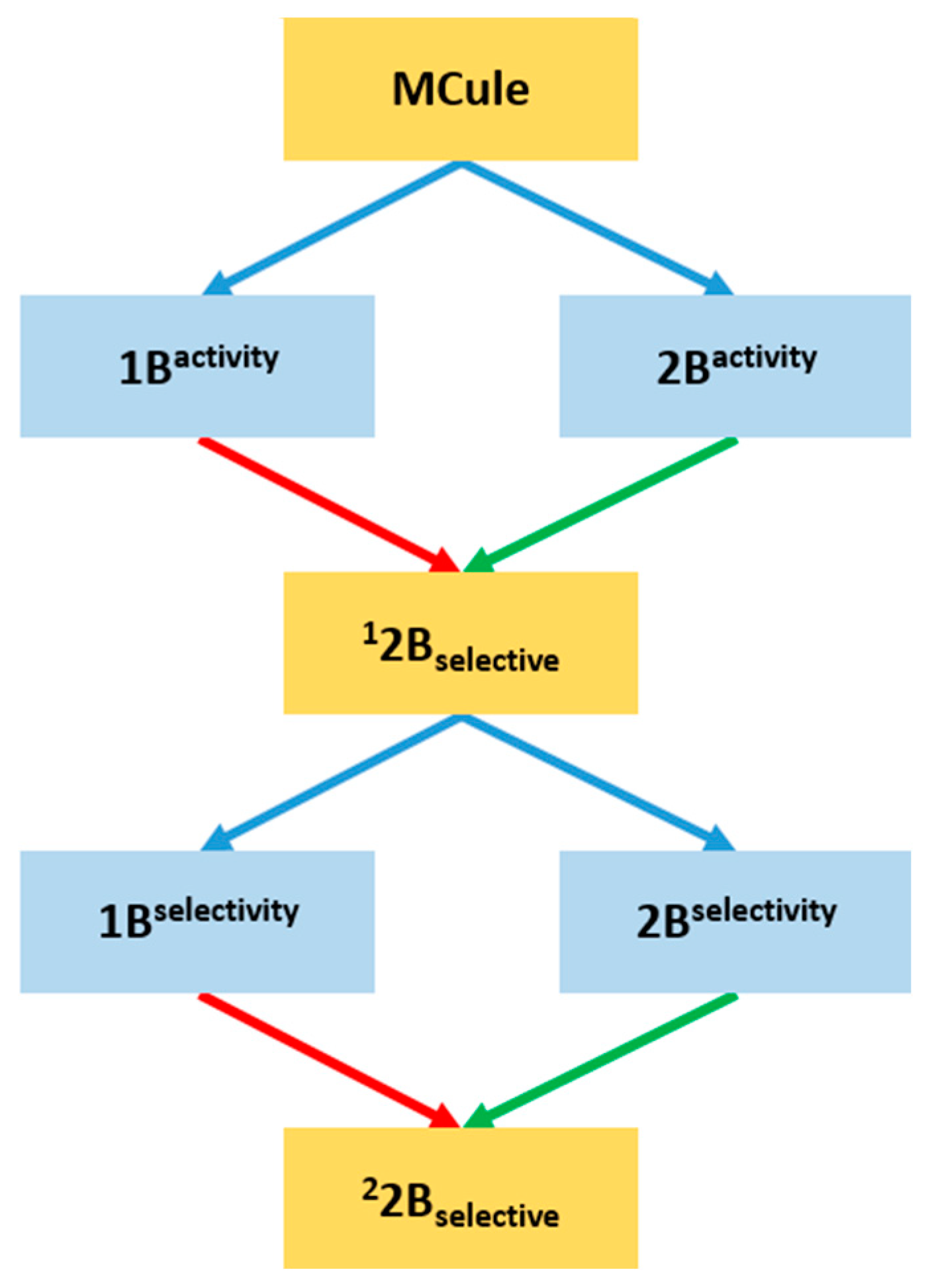
2.1. Compilation of the Training Sets for Machine Learning-Based Classification
2.2. Building NSFP-Based Machine Learning Model
2.3. Prospective Machine Learning-Based Classification
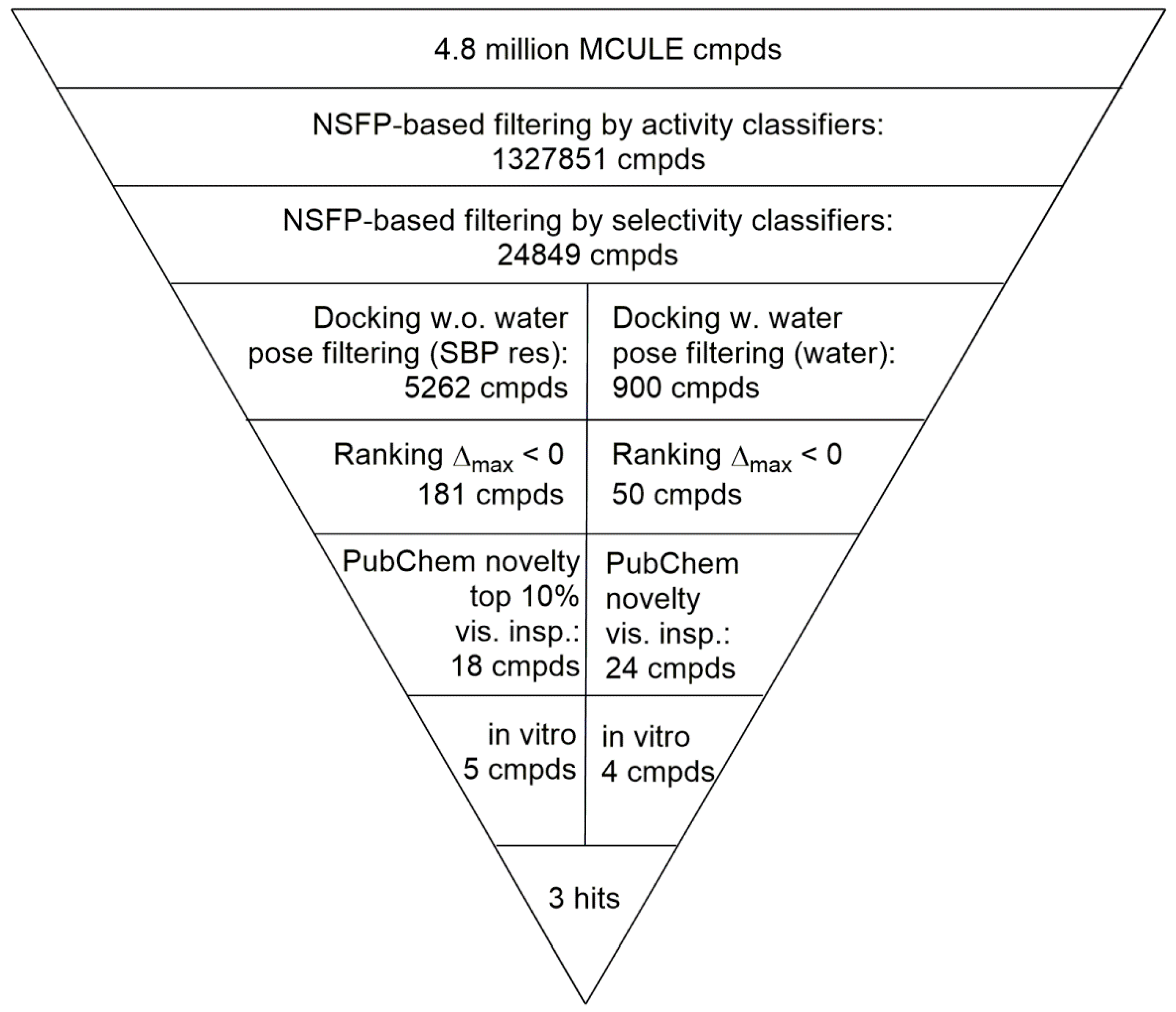
2.4. Virtual Screening of the Prefiltered Set Classified by Machine Learning
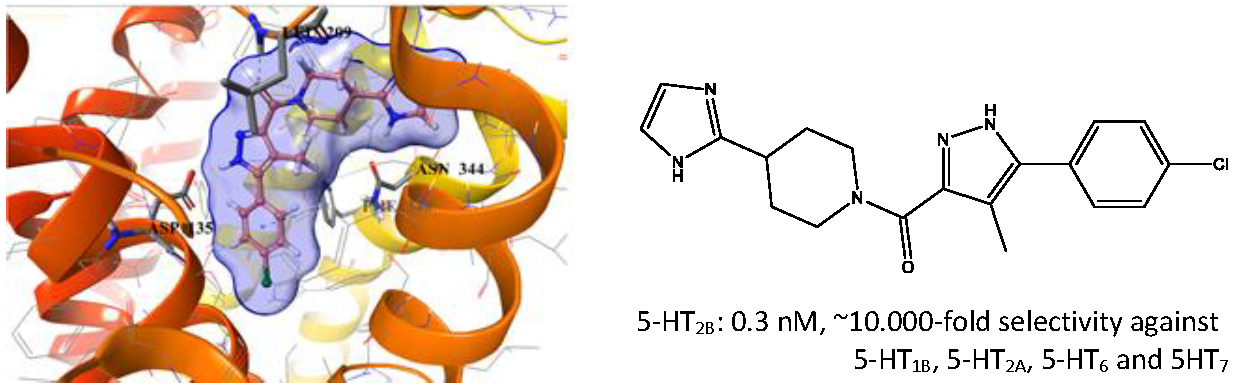
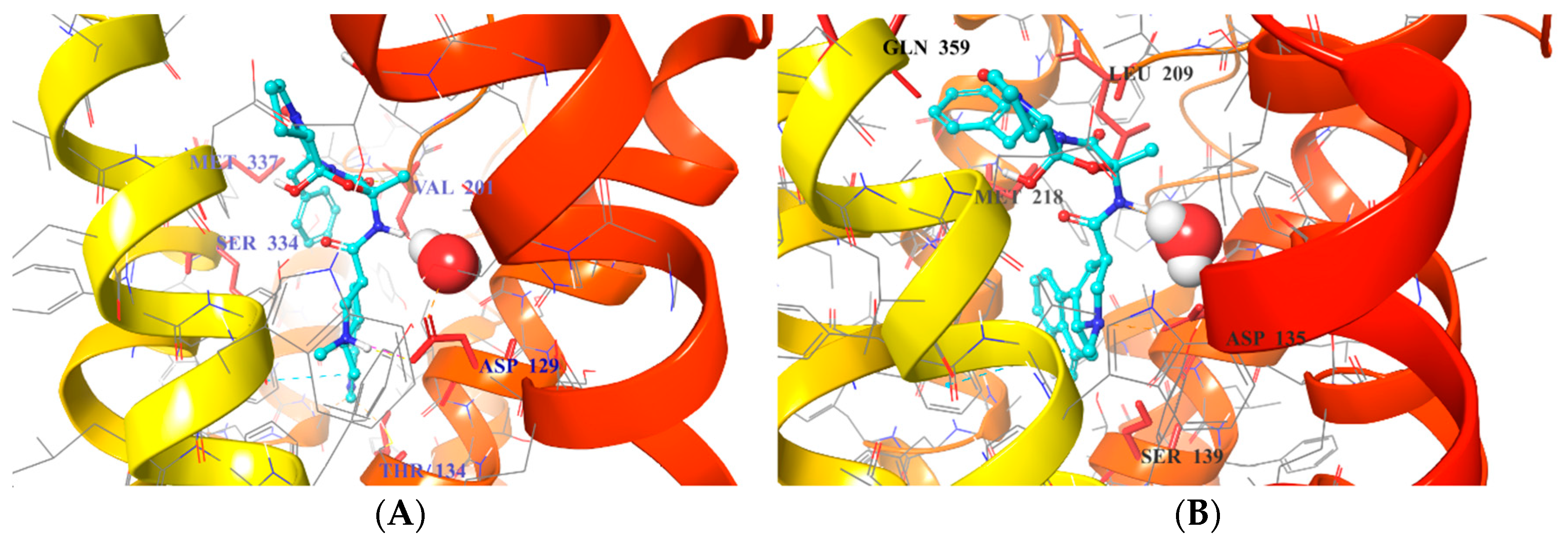
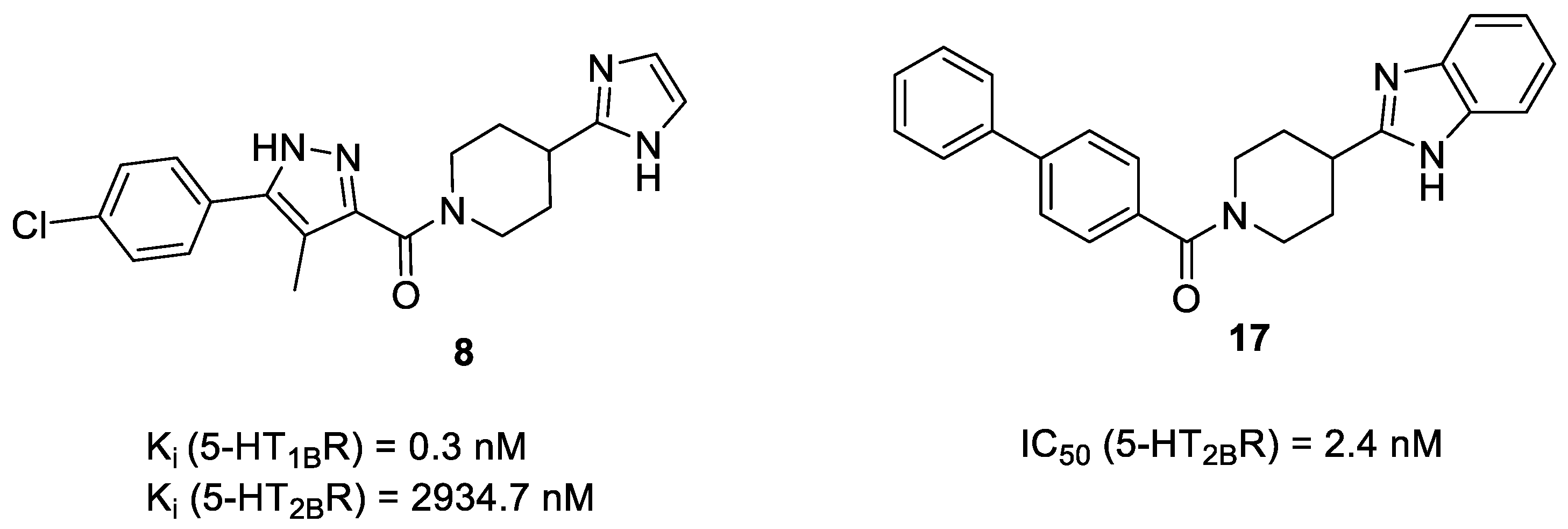
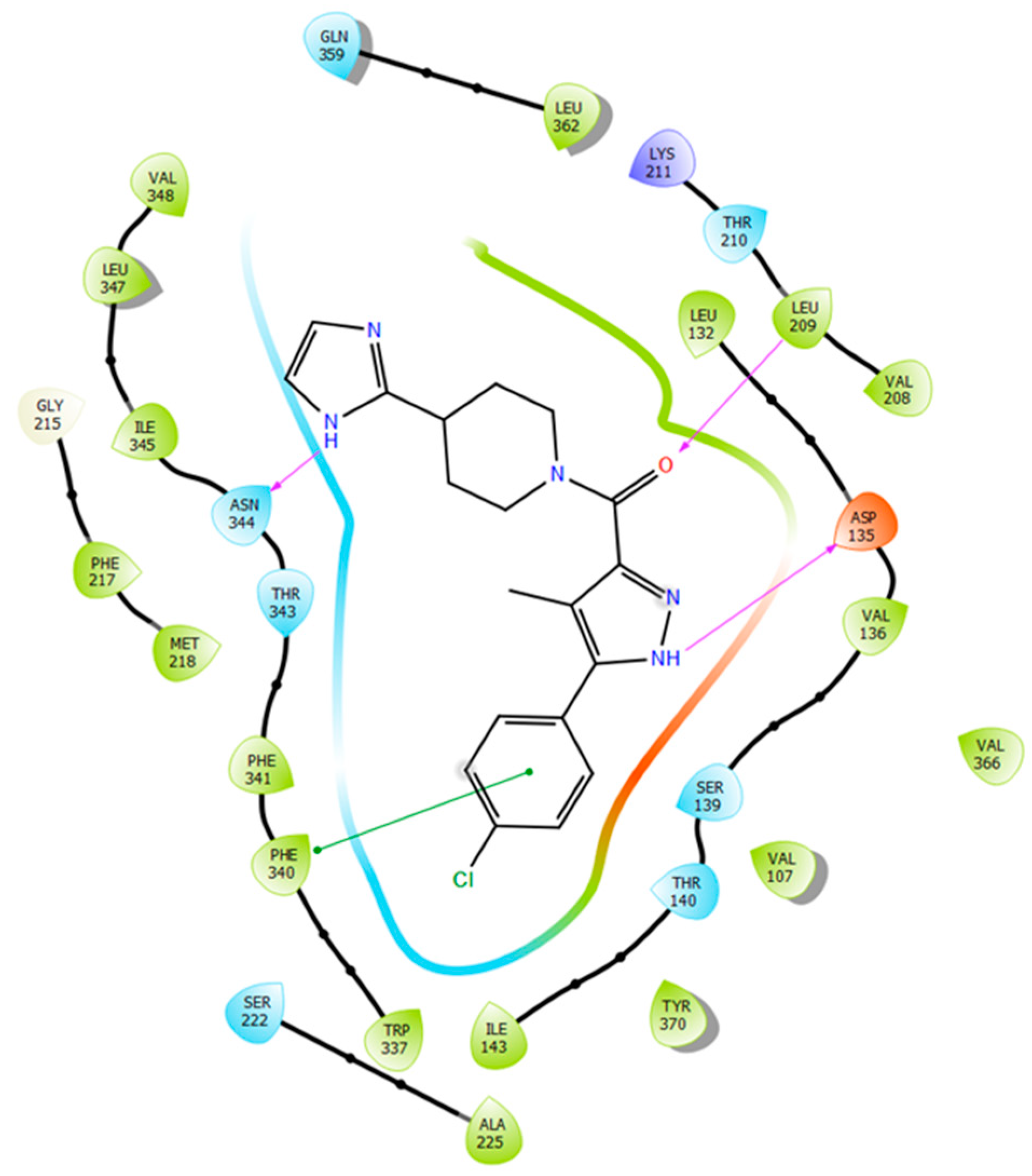
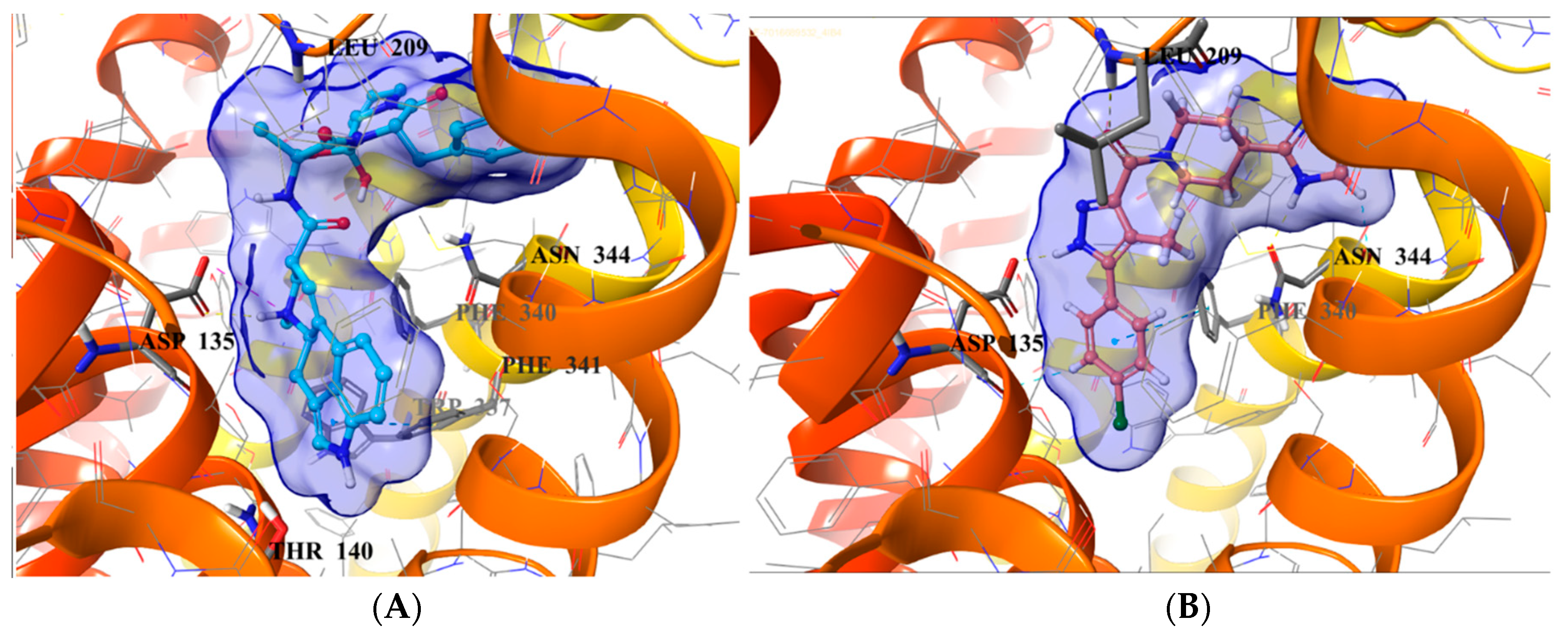
2.5. Binding Mode of Compound 8
3. Conclusions
4. Materials and Methods
4.1. Procedures of Machine Learning-Based Classification Model Building
4.2. Docking-Based Evaluation of NSFP-Classified MCule Sets
4.2.1. Preparation of Ligand Sets, and Receptor Structures for Docking
4.2.2. Evaluation of the Docking Results
4.3. Procedures of In Vitro Screening Assays
4.3.1. Competition Binding in Human 5-HT1B Receptor
4.3.2. Competition Binding in Human 5-HT2B Receptor
4.3.3. Competition Binding in Human 5-HT1A, 5-HT2A 5-HT6 and 5-HT7 Receptors
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Kitson, S.L. 5-hydroxytryptamine (5-HT) receptor ligands. Curr. Pharm. Des. 2007, 13, 2621–2637. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, S.; Siegel, J.; Tikhonova, I.; Jacobson, K. Rhodopsin and the Others: A Historical Perspective on Structural Studies of G Protein-Coupled Receptors. Curr. Pharm. Des. 2009, 15, 3994–4002. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Foon, S.T.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, Y.; Ma, J.; Wu, H.; Wacker, D.; Katritch, V.; Han, G.W.; Liu, W.; Huang, X.-P.; Vardy, E.; et al. Structural basis for molecular recognition at serotonin receptors. Science 2013, 340, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wacker, D.; Gati, C.; Han, G.W.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; Katritch, V.; Barty, A.; et al. Serial Femtosecond Crystallography of G Protein–Coupled Receptors. Science 2013, 342, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E.; et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Rovati, G.E.; Capra, V.; Neubig, R.R.; Smit, M.J.; Roovers, E.; Cotecchia, S.; Leurs, R. The highly conserved DRY motif of class A G protein-coupled receptors: Beyond the ground state. Mol. Pharmacol. 2007, 71, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β2-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.-J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What Can Crystal Structures of Aminergic Receptors Tell Us about Designing Subtype-Selective Ligands? Pharmacol. Rev. 2014, 67, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.W.; Phebus, L.A.; Cohen, M.L. Serotonin in migraine: Theories, animal models and emerging therapies. Prog. Drug Res. 1998, 51, 219–244. [Google Scholar] [CrossRef] [PubMed]
- Fozard, J.R.; Kalkman, H.O. 5-Hydroxytryptamine (5-HT) and the initiation of migraine: New perspectives. Naunyn-Schmiedebergs Arch. Pharmacol. 1994, 350, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, K.; Ullmer, C.; Kalkman, H.O.; Probst, A.; Lübbert, H. Activation of Meningeal 5-HT2B Receptors: An Early Step in the Generation of Migraine Headache? Eur. J. Neurosci. 1996, 8, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Janssen, W.; Schymura, Y.; Novoyatleva, T.; Kojonazarov, B.; Boehm, M.; Wietelmann, A.; Luitel, H.; Murmann, K.; Krompiec, D.R.; Tretyn, A.; et al. 5-HT2B receptor antagonists inhibit fibrosis and protect from RV heart failure. Biomed. Res. Int. 2015, 2015, 438403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ma, J.; Lin, X.; Huang, X.-P.; Wu, K.; Huang, N. Structure-Based Discovery of Novel and Selective 5-Hydroxytryptamine 2B Receptor Antagonists for the Treatment of Irritable Bowel Syndrome. J. Med. Chem. 2016, 59, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Bonhaus, D.W.; Flippin, L.A.; Greenhouse, R.J.; Jaime, S.; Rocha, C.; Dawson, M.; Van Natta, K.; Chang, L.K.; Pulido-Rios, T.; Webber, A.; et al. RS-127445: A selective, high affinity, orally bioavailable 5-HT2B receptor antagonist. Br. J. Pharmacol. 1999, 127, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.R.; Gager, T.L.; Holland, V.; Brown, A.M.; Wood, M.D. m-Chlorophenylpiperazine (mCPP) is an antagonist at the cloned human 5-HT2B receptor. Neuroreport 1996, 7, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.; Gacsályi, I.; Wellmann, J.; Schmidt, É.; Szücs, Z.; Dubreuil, V.; Nicolas, J.P.; Boutin, J.; Bózsing, D.; Egyed, A.; et al. Effects of EGIS-7625, a Selective and Competitive 5-HT2B Receptor Antagonist. Cardiovasc. Drugs Ther. 2003, 17, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Evrard, D.A.; Murdoch, G.R.; Droste, J.J.; Nissen, J.S.; Schenck, K.W.; Fludzinski, P.; Lucaites, V.L.; Nelson, D.L.; Cohen, M.L. Potent, selective tetrahydro-β-carboline antagonists of the serotonin 2B (5HT(2B)) contractile receptor in the rat stomach fundus. J. Med. Chem. 1996, 39, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Forbes, I.T.; Kennett, G.A.; Gadre, A.; Ham, P.; Hayward, C.J.; Martin, R.T.; Thompson, M.; Wood, M.D.; Baxter, G.S. N-(1-Methyl-5-indolyl)-N’-(3-pyridyl)urea hydrochloride: The first selective 5-HT1C receptor antagonist. J. Med. Chem. 1993, 36, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.-E.; Kumar, K.; Wang, X. Predictive In Silico Studies of Human 5-hydroxytryptamine Receptor Subtype 2B (5-HT2B) and Valvular Heart Disease. Curr. Top. Med. Chem. 2013, 13, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Lacivita, E.; Podlewska, S.; Speranza, L.; Niso, M.; Satała, G.; Perrone, R.; Perrone-Capano, C.; Bojarski, A.J.; Leopoldo, M. Structural modifications of the serotonin 5-HT7 receptor agonist N-(4-cyanophenylmethyl)-4-(2-biphenyl)-1-piperazinehexanamide (LP-211) to improve in vitro microsomal stability: A case study. Eur. J. Med. Chem. 2016, 120, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Rataj, K.; Czarnecki, W.; Podlewska, S.; Bojarski, A.J. Substructural Connectivity Fingerprint and Extreme Entropy Machines—A New Method of Compound Representation and Analysis. Molecules 2018. submitted. [Google Scholar]
- Czarnecki, W.M. Weighted Tanimoto Extreme Learning Machine with Case Study in Drug Discovery. IEEE Comput. Intell. Mag. 2015, 10, 19–29. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Klekota, J.; Roth, F.P. Chemical substructures that enrich for biological activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Ágai-Csongor, É.; Horti, F.; Keserű, G.M. Multiple Fragment Docking and Linking in Primary and Secondary Pockets of Dopamine Receptors. ACS Med. Chem. Lett. 2014, 5, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Kristiansen, K.; Kroeze, W.K.; Willins, D.L.; Gelber, E.I.; Savage, J.E.; Glennon, R.A.; Roth, B.L. A highly conserved aspartic acid (Asp-155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT(2A) serotonin receptor but does not participate in activation via a “salt-bridge disruption” mechanism. J. Pharmacol. Exp. Ther. 2000, 293, 735–746. [Google Scholar] [PubMed]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L.; Molinoff, P.B.; Ruffolo, R.R.; Trendelenburg, U. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 2015, 67, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, Á.A.; Kiss, R.; Ferenczy, G.G.; Kovács, L.; Flachner, B.; Lőrincz, Z.; Keserű, G.M. Structure-Based Consensus Scoring Scheme for Selecting Class A Aminergic GPCR Fragments. J. Chem. Inf. Model. 2016, 56, 412–422. [Google Scholar] [CrossRef] [PubMed]
- The PubChem Project. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 24 Janunary 2018).
- Moss, N.; Choi, Y.; Cogan, D.; Flegg, A.; Kahrs, A.; Loke, P.; Meyn, O.; Nagaraja, R.; Napier, S.; Parker, A.; et al. A new class of 5-HT2B antagonists possesses favorable potency, selectivity, and rat pharmacokinetic properties. Bioorgan. Med. Chem. Lett. 2009, 19, 2206–2210. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017.
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-4: Schrödinger Suite 2017-4 Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2017.
- Small-Molecule Drug Discovery Suite 2017-4; Schrödinger, LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-4: Glide; Schrödinger, LLC: New York, NY, USA, 2017.
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
Sample Availability: Samples of the tested compounds are available from commercial vendors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Number of Actives 1 | Number of Inactives 2 | Number of Selectives 3 |
|---|---|---|---|
| 5-HT1B | 858 (1011) | 339 (477) | 86 |
| 5-HT2B | 478 (718) | 259 (351) | 33 |
| Compound | ID | 5-HT1B (%) 1 | 5-HT2B (%) 1 | 5-HT1B (nM) 2 | 5-HT2B (nM) 2 |
|---|---|---|---|---|---|
 | 8a | - | - | 2934.7 ± 321.3 | 0.3 ± 0.07 |
 | 9b | - | - | 2099 ± 622.2 | 235.1 ± 15.9 |
 | 10a | 7 ± 4 | - | - | 2612.9 ± 422.1 |
 | 11a | 12 ± 4 | 40 ± 3 | - | - |
 | 12a | 7 ± 1 | 36 ± 4 | - | - |
 | 13a | 19 ± 3 | 34 ± 4 | - | - |
 | 14b | 26 ± 4 | 34 ± 4 | - | - |
 | 15b | 28 ± 3 | 31 ± 4 | - | - |
 | 16b | 1 ± 1 | 24 ± 4 | - | - |
| Compound | ID | 5-HT1A Ki (µM) | 5-HT2A Ki (µM) | 5-HT6 Ki (µM) | 5-HT7 Ki (µM) |
|---|---|---|---|---|---|
 | 8 | >20 | >20 | 13.2 ± 1.5 | >20 |
 | 9 | >20 | >20 | 7.5 ± 0.9 | >20 |
| Receptor | MCC of Activity Classifiers | MCC of Selectivity Classifiers |
|---|---|---|
| 5-HT1B | 0.7867 | 0.8057 |
| 5-HT2B | 0.7376 | 0.8238 |
| Interactions | 1B Crystals | 2B Crystals |
|---|---|---|
| OBP | D1293.32 | D1353.32 |
| T1343.37 | S1393.36 | |
| S2125.42 | ||
| SBP | Q3597.32 | |
| M3376.58 | M2185.39 | |
| V201ECL2.52 | L209ECL2.52 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rataj, K.; Kelemen, Á.A.; Brea, J.; Loza, M.I.; Bojarski, A.J.; Keserű, G.M. Fingerprint-Based Machine Learning Approach to Identify Potent and Selective 5-HT2BR Ligands. Molecules 2018, 23, 1137. https://doi.org/10.3390/molecules23051137
Rataj K, Kelemen ÁA, Brea J, Loza MI, Bojarski AJ, Keserű GM. Fingerprint-Based Machine Learning Approach to Identify Potent and Selective 5-HT2BR Ligands. Molecules. 2018; 23(5):1137. https://doi.org/10.3390/molecules23051137
Chicago/Turabian StyleRataj, Krzysztof, Ádám Andor Kelemen, José Brea, María Isabel Loza, Andrzej J. Bojarski, and György Miklós Keserű. 2018. "Fingerprint-Based Machine Learning Approach to Identify Potent and Selective 5-HT2BR Ligands" Molecules 23, no. 5: 1137. https://doi.org/10.3390/molecules23051137
APA StyleRataj, K., Kelemen, Á. A., Brea, J., Loza, M. I., Bojarski, A. J., & Keserű, G. M. (2018). Fingerprint-Based Machine Learning Approach to Identify Potent and Selective 5-HT2BR Ligands. Molecules, 23(5), 1137. https://doi.org/10.3390/molecules23051137