
Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides


Abstract
:1. Introduction
Challenges in IP Chemotherapy
2. Affinity Targeting of Tumors with Homing Peptides
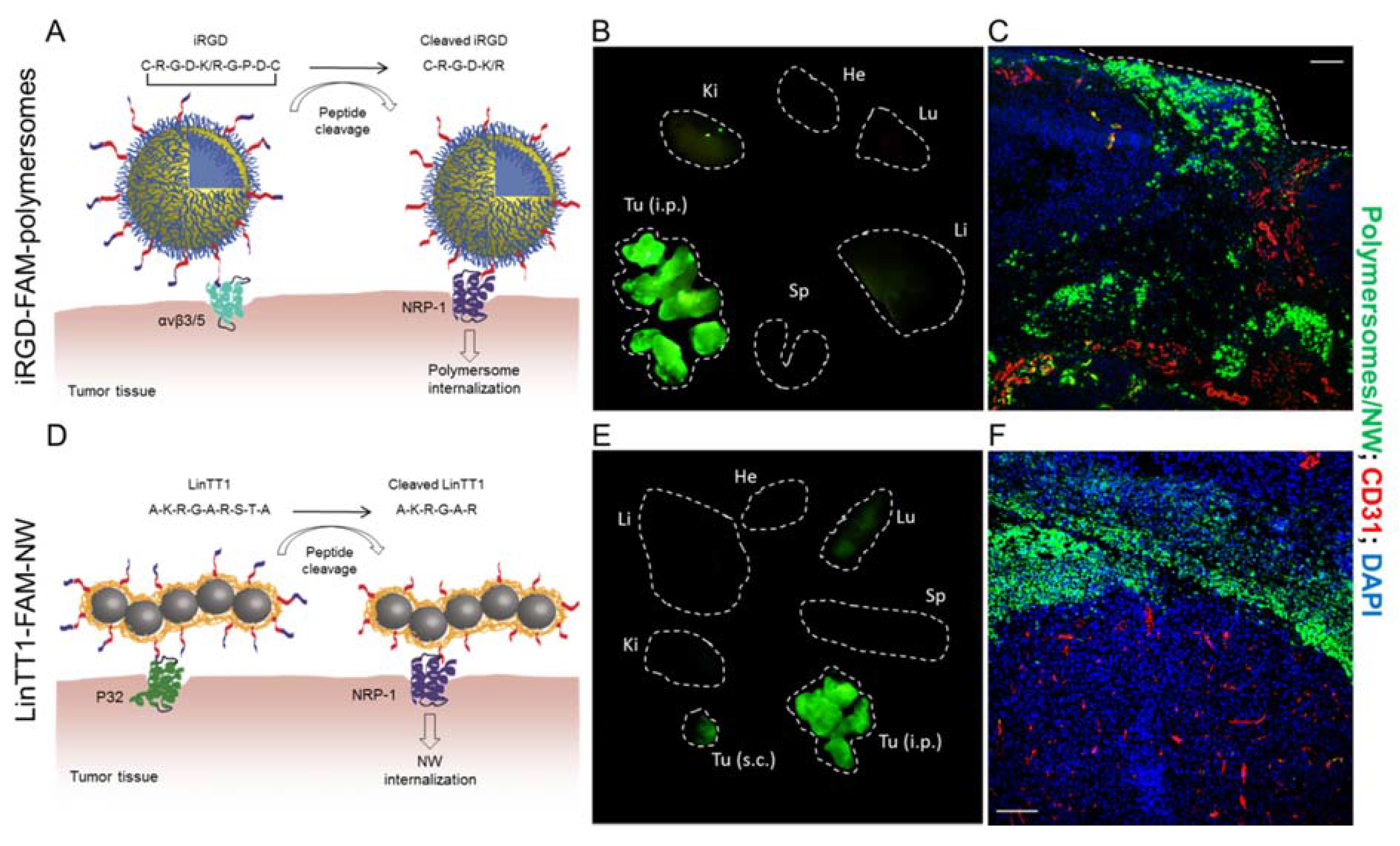
2.1. Integrin Targeting Peptides
2.2. Tumor-Penetrating Peptides
2.3. M2 Macrophage-Targeting Peptide
2.4. Nucleolin Targeting Peptide
2.5. EphA2 Targeting Peptide
2.6. Hyaluronan Targeting Peptide
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Coccolini, F.; Gheza, F.; Lotti, M.; Virzì, S.; Iusco, D.; Ghermandi, C.; Melotti, R.; Baiocchi, G.; Giulini, S.M.; Ansaloni, L.; et al. Peritoneal carcinomatosis. World J. Gastroenterol. 2013, 19, 6979–6994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugarbaker, P.H. Overview of peritoneal carcinomatosis. Cancerologia 2008, 3, 119–124. [Google Scholar]
- Goodman, M.D.; McPartland, S.; Detelich, D.; Saif, M.W. Chemotherapy for intraperitoneal use: A review of hyperthermic intraperitoneal chemotherapy and early post-operative intraperitoneal chemotherapy. J. Gastrointest. Oncol. 2016, 7, 45–57. [Google Scholar] [PubMed]
- Klaver, Y.L.; Lemmens, V.E.; Nienhuijs, S.W.; Luyer, M.D.; de Hingh, I.H. Peritoneal carcinomatosis of colorectal origin: Incidence, prognosis and treatment options. World J. Gastroenterol. 2012, 18, 5489–5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Venegas, R.; Fey, R.; Van Der Heyde, H.; Bernard, M.A.; Lazarides, E.; Woods, C.M. An in vivo approach to structure activity relationship analysis of peptide ligands. Pharm. Res. 2007, 24, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Franko, J.; Ibrahim, Z.; Gusani, N.J.; Holtzman, M.P.; Bartlett, D.L.; Zeh, H.J. Cytoreductive surgery and hyperthermic intraperitoneal chemoperfusion versus systemic chemotherapy alone for colorectal peritoneal carcinomatosis. Cancer 2010, 116, 3756–3762. [Google Scholar] [CrossRef] [PubMed]
- Lambert, L.A. Looking up: Recent advances in understanding and treating peritoneal carcinomatosis. CA Cancer J. Clin. 2015, 65, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wang, J.; Wientjes, M.G.; Au, J.L.-S. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010, 6, 1625–1641. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.L.; Myers, C.E.; Bungay, P.M.; DeVita, V.T. Pharmacokinetic rationale for peritoneal drug administration in the treatment of ovarian cancer. Cancer Treat. Rep. 1978, 62, 1–13. [Google Scholar] [PubMed]
- Bajaj, G.; Yeo, Y. Drug delivery systems for intraperitoneal therapy. Pharm. Res. 2010, 27, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, J. Intraperitoneal chemotherapy against peritoneal carcinomatosis. Surg. Oncol. 2014, 23, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Brcher, B.L.; Piso, P.; Verwaal, V.; Esquivel, J.; Derraco, M.; Yonemura, Y.; Gonzalez-Moreno, S.; Pelz, J.; Königsrainer, A.; Ströhlein, M.; et al. Peritoneal carcinomatosis: Cytoreductive surgery and hipec-overview and basics. Cancer Investig. 2012, 30, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Girshally, R.; Demtröder, C.; Albayrak, N.; Zieren, J.; Tempfer, C.; Reymond, M.A. Pressurized intraperitoneal aerosol chemotherapy (PIPAC) as a neoadjuvant therapy before cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. World J. Surg. Oncol. 2016, 14, 253. [Google Scholar] [CrossRef] [PubMed]
- Flessner, M.F.; Fenstermacher, J.D.; Blasberg, R.G.; Dedrick, R.L. Peritoneal absorption of macromolecules studied by quantitative autoradiography. Am. J. Physiol. 1985, 248, H26–H32. [Google Scholar] [CrossRef] [PubMed]
- Hlrano, K.; Anthony Hunt, C. Lymphatic transport of liposome-encapsulated agents: Effects of liposome size following intraperitoneal administration. J. Pharm. Sci. 1985, 74, 915–921. [Google Scholar]
- Poveda, A.; Salazar, R.; del Campo, J.M.; Mendiola, C.; Cassinello, J.; Ojeda, B.; Arranz, J.A.; Oaknin, A.; García-Foncillas, J.; Rubio, M.J.; et al. Update in the management of ovarian and cervical carcinoma. Clin. Transl. Oncol. 2007, 9, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dakwar, G.R.; Shariati, M.; Willaert, W.; Ceelen, W.; De Smedt, S.C.; Remaut, K. Nanomedicine-Based intraperitoneal therapy for the treatment of peritoneal carcinomatosis—mission possible? Adv. Drug Deliv. Rev. 2017, 108, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Bennis, S.; Chapey, C.; Robert, J.; Couvreur, P. Enhanced cytotoxicity of doxorubicin encapsulated in polyisohexylcyanoacrylate nanospheres against multidrug-resistant tumour cells in culture. Eur. J. Cancer 1994, 30, 89–93. [Google Scholar] [CrossRef]
- Goren, D.; Horowitz, A.T.; Tzemach, D.; Tarshish, M.; Zalipsky, S.; Gabizon, A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin. Cancer Res. 2000, 6, 1949–1957. [Google Scholar] [PubMed]
- Sadava, D.; Coleman, A.; Kane, S.E. Liposomal daunorubicin overcomes drug resistance in human breast, ovarian and lung carcinoma cells. J. Liposome Res. 2002, 12, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Morris, C.Q. Antibody-Drug Conjugates (ADCs) for personalized treatment of solid tumors: A review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Braun, G.B.; Luo, X.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. Application of a proapoptotic peptide to intratumorally spreading cancer therapy. Cancer Res. 2013, 73, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Qin, Y.; Liu, H.; Cheng, Z. Tumor-Targeting Peptides: Ligands for molecular imaging and therapy. Anticancer Agents Med. Chem. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; Kruger, H.G. Therapeutic peptides. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Paradís-Bas, M.; Tulla-Puche, J.; Albericio, F. The road to the synthesis of “difficult Peptides”. Chem. Soc. Rev. 2016, 45, 631–654. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Mapping of vascular zip codes by phage display. Methods Enzymol. 2012, 503, 35–56. [Google Scholar] [PubMed]
- Ruoslahti, E. Specialization of tumour vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Tumor-penetrating peptides. Front. Oncol. 2013, 3, 216. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, H.; Lingasamy, P.; Anton Willmore, A.-M.; Hunt, H.; Kurm, K.; Tammik, O.; Scodeller, P.; Simón-Gracia, L.; Kotamraju, V.R.; Lowy, A.M.; et al. Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Akita, N.; Maruta, F.; Seymour, L.W.; Kerr, D.J.; Parker, A.L.; Asai, T.; Oku, N.; Nakayama, J.; Miyagawa, S. Identification of oligopeptides binding to peritoneal tumors of gastric cancer. Cancer Sci. 2006, 97, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Hamidi, H.; Pietilä, M.; Ivaska, J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer 2016, 115, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.-J.; Schwaiger, M.; Weinmüller, M.; Räder, A.; Steiger, K.; Kessler, H. Exploring the role of rgd-recognizing integrins in cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Scodeller, P.; Braun, G.B.; De Mendoza, T.H.; Yamazaki, C.M.; Kluger, M.D.; Kitayama, J.; Alvarez, E.; Howell, S.B.; Teesalu, T.; et al. A Tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J. Control. Release 2015, 212, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Simón-Gracia, L.; Hunt, H.; Scodeller, P.; Gaitzsch, J.; Kotamraju, V.R.; Sugahara, K.N.; Tammik, O.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 2016, 104, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A.; Ottensmeier, C.; Niloff, J.; Orta, B.; DiCarlo, J. Expression and Function of β1 and αvβ3 integrins in ovarian cancer. Gynecol. Oncol. 1995, 58, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Kruijtzer, J.A.W.; Frielink, C.; Corstens, F.H.M.; Oyen, W.J.G.; Liskamp, R.M.J.; Boerman, O.C. αvβ3 Integrin-targeting of intraperitoneally growing tumors with a radiolabeled RGD peptide. Int. J. Cancer 2007, 120, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.-H.; Josserand, V.; Razkin, J.; Garanger, E.; Boturyn, D.; Favrot, M.-C.; Dumy, P.; Coll, J.-L. Noninvasive optical imaging of ovarian metastases using Cy5-Labeled RAFT-c(-RGDfK-)4. Mol. Imaging 2006, 5, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Keramidas, M.; Josserand, V.; Righini, C.A.; Wenk, C.; Faure, C.; Coll, J.L. Intraoperative near-infrared image-guided surgery for peritoneal carcinomatosis in a preclinical experimental Model. Br. J. Surg. 2010, 97, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Chi, C.; Shang, W.; Rengaowa, S.; Cui, J.; Ye, J.; Jiang, S.; Mao, Y.; Zeng, C.; Huo, H.; et al. Precise integrin-targeting near-infrared imaging-guided surgical method increases surgical qualification of peritoneal carcino-matosis from gastric cancer in mice. Oncotarget 2016, 8, 6258–6272. [Google Scholar]
- Sluiter, N.; de Cuba, E.; Kwakman, R.; Kazemier, G.; Meijer, G.; te Velde, E.A. Adhesion molecules in peritoneal dissemination: Function, prognostic relevance and therapeutic options. Clin. Exp. Metastasis 2016, 33, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Pierschbacher, M.D. Arg-Gly-Asp: A versatile cell recognition signal. Cell 1986, 44, 517–518. [Google Scholar] [CrossRef]
- Graf, J.; Iwamoto, Y.; Sasaki, M.; Martin, G.R.; Kleinman, H.K.; Robey, F.A.; Yamada, Y. Identification of an amino acid sequence in laminin mediating cell attachment, chemotaxis, and receptor binding. Cell 1987, 48, 989–996. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Robey, F.A.; Graf, J.; Sasaki, M.; Kleinman, H.K.; Yamada, Y.; Martin, G.R. YIGSR, a synthetic laminin pentapeptide, inhibits experimental metastasis formation. Science 1987, 238, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Braun, G.B.; de Mendoza, T.H.; Kotamraju, V.R.; French, R.P.; Lowy, A.M.; Teesalu, T.; Ruoslahti, E. Tumor-penetrating iRGD peptide inhibits metastasis. Mol. Cancer Ther. 2015, 14, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Hirakawa-YS Chung, K.; Yashiro, M.; Nishimura, S.; Sawada, T.; Saiki, I.; Sowa, M. Adhesion polypeptides are useful for the prevention of peritoneal dissemination of gastric cancer. Clin. Exp. Metastasis 1997, 16, 381–388. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Yamaguchi, K.; Shiraishi, N.; Adachi, Y.; Saiki, I.; Kitano, S. Port-Site metastasis after CO2 pneumoperitoneum. Surg. Endosc. 2004, 18, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Zanuy, D.; Kotla, R.; Nussinov, R.; Teesalu, T.; Sugahara, K.N.; Alemán, C.; Haspel, N. Sequence dependence of c-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 2013, 182, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-End Rule Peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed]
- Braun, G.B.; Sugahara, K.N.; Yu, O.M.; Kotamraju, V.R.; Mölder, T.; Lowy, A.M.; Ruoslahti, E.; Teesalu, T. Urokinase-controlled tumor penetrating peptide. J. Control. Release 2016, 232, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tam, S.; Ingham, E.S.; Mahakian, L.M.; Lai, C.Y.; Tumbale, S.K.; Teesalu, T.; Hubbard, N.E.; Borowsky, A.D.; Ferrara, K.W. Ultrasound molecular imaging of tumor angiogenesis with a neuropilin-1-targeted microbubble. Biomaterials 2015, 56, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Pang, H.-B.; Braun, G.B.; Friman, T.; Aza-Blanc, P.; Ruidiaz, M.E.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. An Endocytosis Pathway Initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 2014, 5, 4904. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptdei enhances the efficacy of cancer drugs. Science 2010, 328, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Simón-Gracia, L.; Hunt, H.; Scodeller, P.D.; Gaitzsch, J.; Braun, G.B.; Willmore, A.-M.A.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. Paclitaxel-loaded polymersomes for enhanced intraperitoneal chemotherapy. Mol. Cancer Ther. 2016, 15, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Wonder, E.; Simón-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.K.; Teesalu, T.; Safinya, C.R. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome–DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Paasonen, L.; Sharma, S.; Braun, G.B.; Kotamraju, V.R.; Chung, T.D.Y.; She, Z.-G.; Sugahara, K.N.; Yliperttula, M.; Wu, B.; Pellecchia, M.; et al. New p32/gC1qR ligands for targeted tumor drug delivery. Chembiochem 2016, 17, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kotamraju, V.R.; Mölder, T.; Tobi, A.; Teesalu, T.; Ruoslahti, E. Tumor-Penetrating nanosystem strongly suppresses breast tumor growth. Nano Lett. 2017, 17, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Hunt, H.; Simón-Gracia, L.; Tobi, A.; Kotamraju, V.R.; Sharma, S.; Nigul, M.; Sugahara, K.N.; Ruoslahti, E.; Teesalu, T. Targeting of p32 in peritoneal carcinomatosis with intraperitoneal linTT1 peptide-guided pro-apoptotic nanoparticles. J. Control. Release 2017, 260, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Simon-Gracia, L.; Scodeller, P.; Di Silvio, D.; Salzar Fuentes, S.; Gomez Vallejo, V.; Rios, X.; San Sebastian, E.; Suck, M.; De Lorenzi, F.; Yokota Rizzo, L.; et al. Detection of small breast tumors using tumor penetrating-polymersomes engineered to target p32 protein. bioRxiv 2017. [Google Scholar] [CrossRef]
- Fogal, V.; Zhang, L.; Krajewski, S.; Ruoslahti, E. Mitochondrial/Cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008, 68, 7210–7218. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Akerman, M.E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R.M.; Ruoslahti, E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9381–9386. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Fushida, S.; Yamamoto, Y.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Miyashita, T.; Tajima, H.; Ninomiya, I.; Munesue, S.; et al. Tumor-associated macrophages of the m2 phenotype contribute to progression in gastric cancer with peritoneal dissemination. Gastric Cancer 2016, 19, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Scodeller, P.; Simón-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Säälik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision targeting of tumor macrophages with a CD206 binding peptide. Sci. Rep. 2017, 7, 14655. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, V.V.; Gillies, R.J. Janus-Faced tumor microenvironment and redox. Antioxid. Redox Signal. 2014, 21, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Porkka, K.; Laakkonen, P.; Hoffman, J.A.; Bernasconi, M.; Ruoslahti, E. A fragment of the hmgn2 protein homes to the nuclei of tumor cells and tumor endothelial cells in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7444–7449. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, S.; Hock, R.; Ueda, T.; Catez, F.; Rochman, M.; Bustin, M. Cell Cycle-dependent binding of HMGN proteins to chromatin. Mol. Biol. Cell 2008, 19, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.; Pilch, J.; Akerman, M.E.; Porkka, K.; Laakkonen, P.; Ruoslahti, E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 2003, 163, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Pollard, H.B. Molecular dissection of nucleolin’s role in growth and cell proliferation: New Insights. FASEB J. 1999, 13, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Drecoll, E.; Gaertner, F.C.; Miederer, M.; Blechert, B.; Vallon, M.; Müller, J.M.; Alke, A.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; et al. Treatment of peritoneal carcinomatosis by targeted delivery of the radio-labeled tumor homing peptide 213Bi-DTPA-[F3]2 into the nucleus of tumor cells. PLoS ONE 2009, 4, e5715. [Google Scholar] [CrossRef] [PubMed]
- Essler, M.; Gärtner, F.C.; Neff, F.; Blechert, B.; Senekowitsch-Schmidtke, R.; Bruchertseifer, F.; Morgenstern, A.; Seidl, C. Therapeutic efficacy and toxicity of 225Ac-Labelled vs. 213Bi-labelled tumour-homing peptides in a preclinical mouse model of peritoneal carcinomatosis. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Vallon, M.; Seidl, C.; Blechert, B.; Li, Z.; Gilbertz, K.-P.; Baumgart, A.; Aichler, M.; Feuchtinger, A.; Gaertner, F.C.; Bruchertseifer, F.; et al. Enhanced efficacy of combined 213Bi-DTPA-F3 and paclitaxel therapy of peritoneal carcinomatosis is mediated by enhanced induction of apoptosis and G2/M phase arrest. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Dong, Z.; Qiao, Y.; Kristensen, G.B.; Holm, R.; Nesland, J.M.; Suo, Z. The clinical significance of epha2 and ephrin a-1 in epithelial ovarian carcinomas. Gynecol. Oncol. 2005, 99, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Deavers, M.; Celestino, J.; Thornton, A.; Fletcher, M.S.; Landen, C.N.; Kinch, M.S.; Kiener, P.A.; Sood, A.K. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin. Cancer Res. 2004, 10, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Koolpe, M.; Dail, M.; Pasquale, E.B. An ephrin mimetic peptide that selectively targets the epha2 receptor. J. Biol. Chem. 2002, 277, 46974–46979. [Google Scholar] [CrossRef] [PubMed]
- Scarberry, K.E.; Dickerson, E.B.; McDonald, J.F.; Zhang, Z.J. Magnetic nanoparticle−peptide conjugates for in vitro and in vivo targeting and extraction of cancer cells. J. Am. Chem. Soc. 2008, 130, 10258–10262. [Google Scholar] [CrossRef] [PubMed]
- Scarberry, K.E.; Dickerson, E.B.; Zhang, Z.J.; Benigno, B.B.; McDonald, J.F. Selective removal of ovarian cancer cells from human ascites fluid using magnetic nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Ween, M.P.; Oehler, M.K.; Ricciardelli, C. Role of versican, hyaluronan and cd44 in ovarian cancer metastasis. Int. J. Mol. Sci. 2011, 12, 1009–1029. [Google Scholar] [CrossRef] [PubMed]
- Willmore, A.-M.; Simón-Gracia, L.; Toome, K.; Paiste, P.; Kotamraju, V.R.; Mölder, T.; Sugahara, K.N.; Ruoslahti, E.; Braun, G.B.; Teesalu, T. Targeted silver nanoparticles for ratiometric cell phenotyping. Nanoscale 2016, 8, 9096–9101. [Google Scholar] [CrossRef] [PubMed]
- Braun, G.B.; Friman, T.; Pang, H.B.; Pallaoro, A.; De Mendoza, T.H.; Willmore, A.M.A.; Kotamraju, V.R.; Mann, A.P.; She, Z.G.; Sugahara, K.N.; et al. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 2014, 13, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Braun, G.B.; Qin, M.; Ruoslahti, E.; Sugahara, K.N. In vivo cation exchange in quantum dots for tumor-specific imaging. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Targeting Peptide | Cargo (Drug or Imaging Molecule) | Target | Animal Tumor Model | Application | Outcome | References |
|---|---|---|---|---|---|---|
| c(RGDfK) | DOTA (tetraxetan)-111In/177Lu | αvβ3/5 | OVCAR-3 | Tumor treatment | Significant increase in survival. | [42] |
| Cy5 dye | Integrin | IGROV1 | Guided tumor resection | Detection of 1-to 5-mm IP tumor nodules. | [43] | |
| Alexa Fluor700 | TSA-pGL3 | Guided tumor resection | 2-fold increase in sensitivity detection of tumors; surgery time reduced from 20 to 14 min. | [44] | ||
| Indocyanine green | SGC-7901 | Guided tumor resection | Detection of 1.8 mm tumors; the operative time was shortened by 3-fold. | [45] | ||
| iRGD (CRGDKGPDC) | Fluorescein; Doxorubicin | αvβ3/5 Integrin/NRP-1 | MKN45P; LOVO-6; IGROV-1 | PC treatment and imaging | 250% more DOX accumulation in tumor; significant tumor growth reduction in MKN45P model. | [39] |
| PTX-loaded polymersomes | MKN45P; CT26 | PC treatment and imaging | Significant tumor growth reduction in MKN45P model. | [40] | ||
| KLP (SWKLPPS) | Adriamycin-encapsulated liposomes | α3β1 Integrin | AZ-P7a | Significantly higher binding to peritoneal tumors compared with control liposomes. | [34] | |
| LinTT1 (AKRGARSTA) | Apoptotic peptide-iron oxide nanoworms | P32/gC1qR | MKN45P; CT26; SKOV-3 | PC treatment and imaging | Significant tumor growth reduction in MKN45P. | [65] |
| IP3 (CKRDLSRRC) | Fluorescein; Silver NP | Hyaluronic acid | MKN45P; CT26 | Specific IP tumor target and penetration. | [33] | |
| UNO (CSPGAKVRC) | Fluorescein; polymersomes | CD206/MRC1 | MKN45P | Specific targeting of M2 macrophages in peritoneal tumors. | [74] | |
| F3 (KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK) | 213Bi; 225Ac | Nucleolin | MDA-MB-435S OVCAR-3 | PC treatment | Significant survival increase; decrease of the number of peritoneal tumors. | [81,82,83] |
| 213Bi, combined with PTX | PC treatment | Significant survival increase but not complete remission. | ||||
| YSA (YSAYPDSVPMMS) | Magnetic nanoparticles | EphA2 | Hey | PC treatment | Removal of tumor cells from IP cavity. | [87] |
| Removal of ovarian cancer cells from ascites in vitro. | [88] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simón-Gracia, L.; Hunt, H.; Teesalu, T. Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides. Molecules 2018, 23, 1190. https://doi.org/10.3390/molecules23051190
Simón-Gracia L, Hunt H, Teesalu T. Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides. Molecules. 2018; 23(5):1190. https://doi.org/10.3390/molecules23051190
Chicago/Turabian StyleSimón-Gracia, Lorena, Hedi Hunt, and Tambet Teesalu. 2018. "Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides" Molecules 23, no. 5: 1190. https://doi.org/10.3390/molecules23051190
APA StyleSimón-Gracia, L., Hunt, H., & Teesalu, T. (2018). Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides. Molecules, 23(5), 1190. https://doi.org/10.3390/molecules23051190