Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles


Abstract
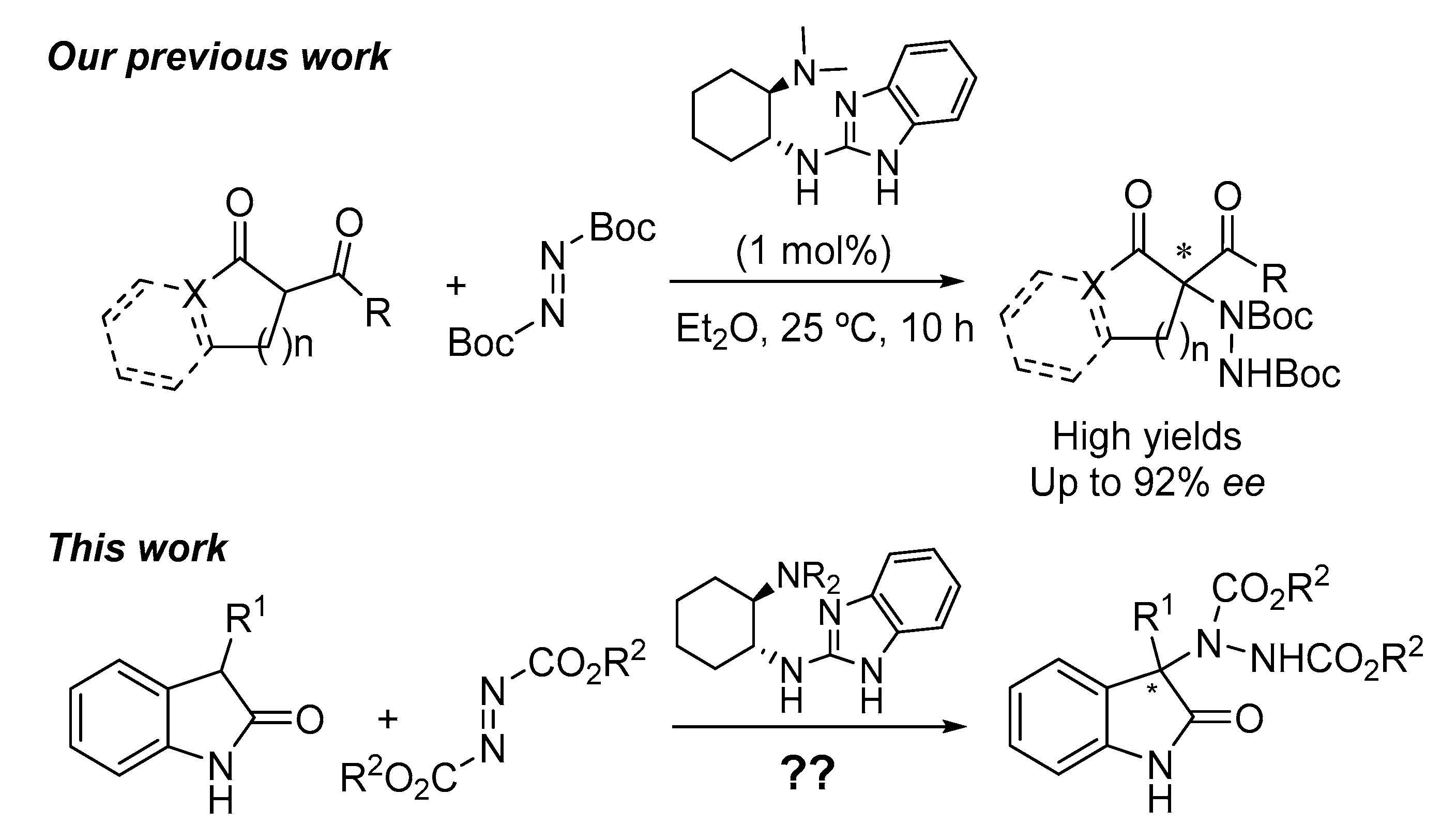
:1. Introduction
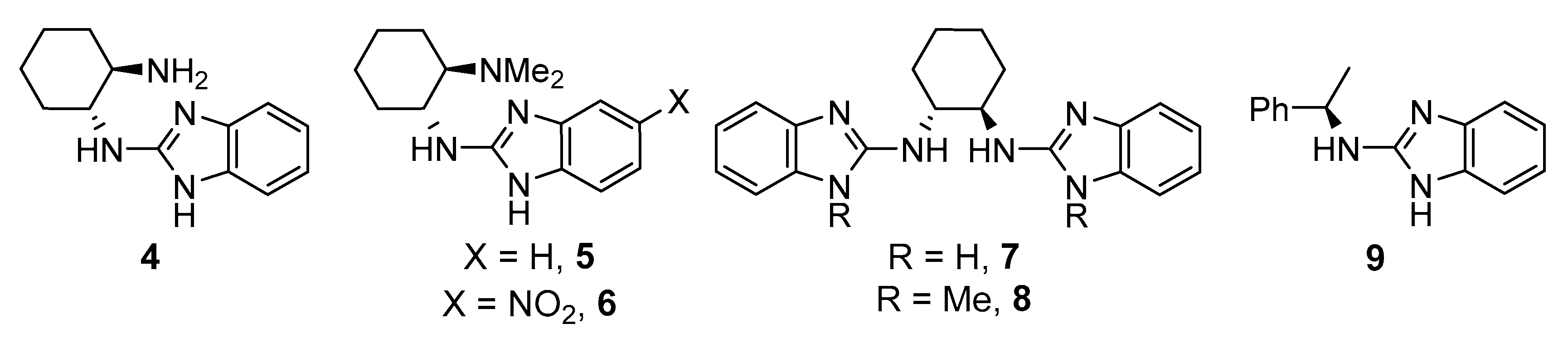
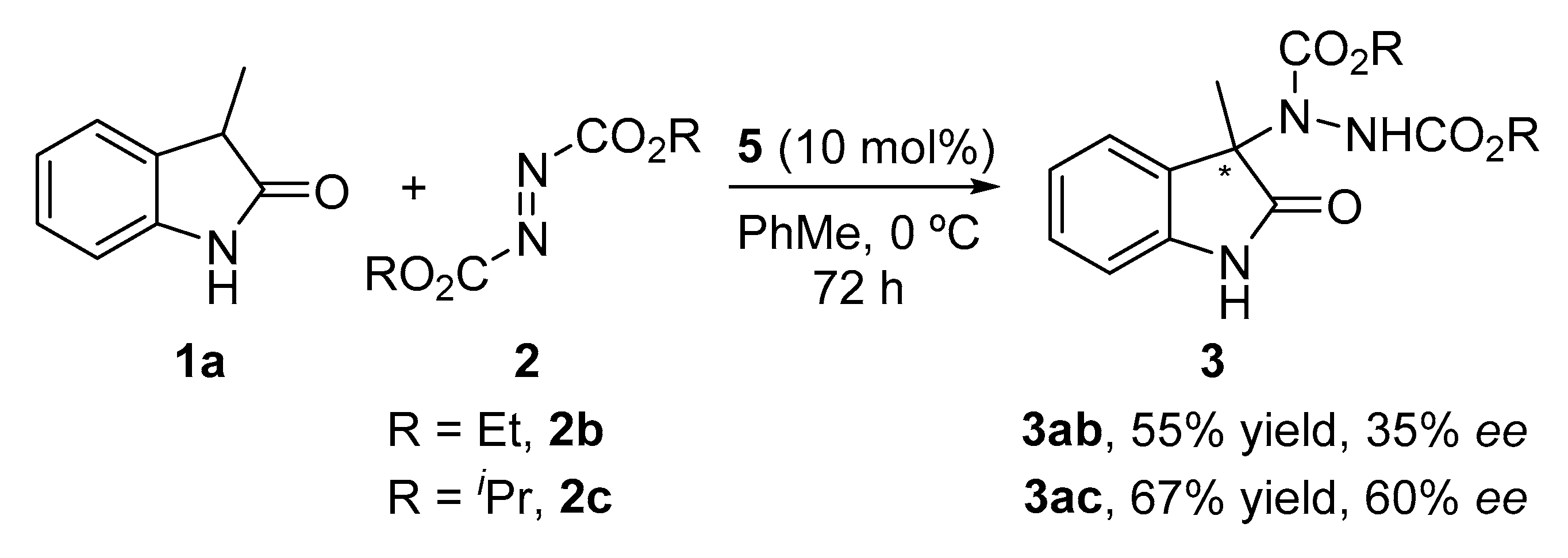
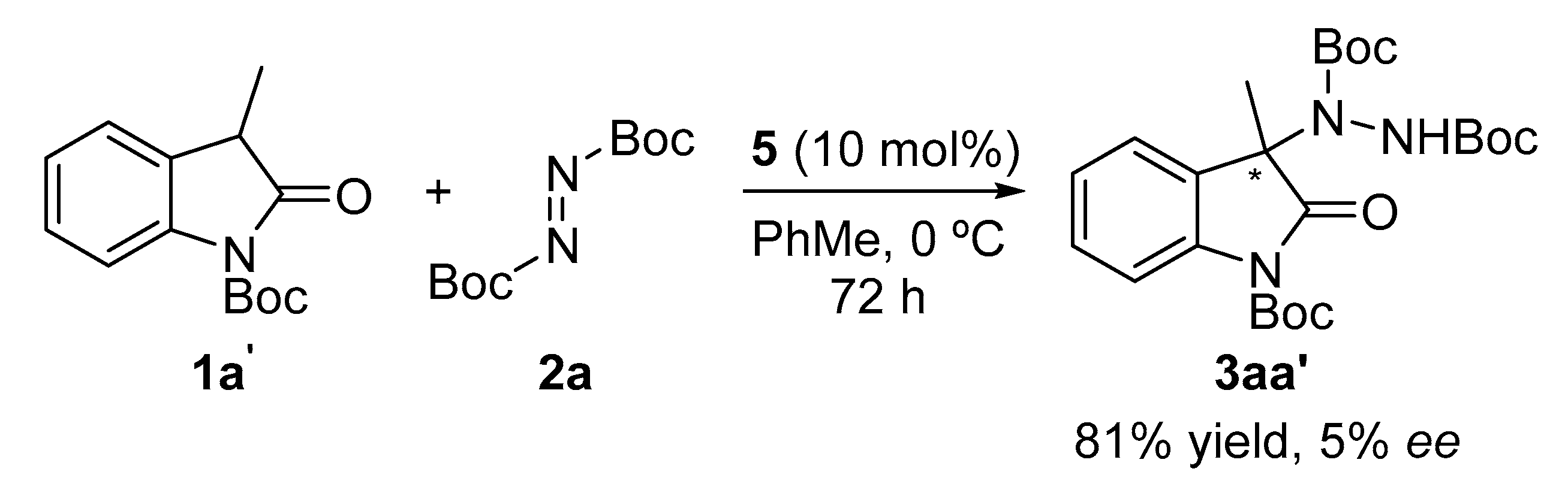
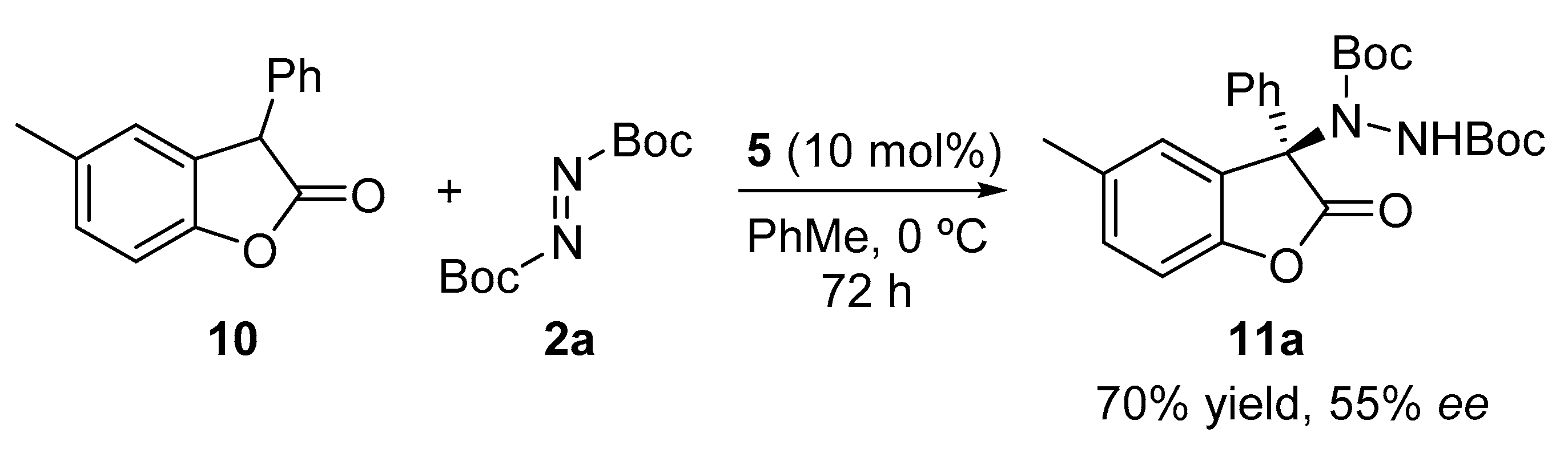
2. Results and Discussion
3. Materials and Methods
General Procedure for the Asymmetric Amination of 3-Substituted Oxindoles
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Barluenga, J.; Valdés, C. Five-membered heterocycles: Indole and related systems. In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH Verlag & Co.: Weinheim, Germany, 2011; Volume 1, pp. 495–498. [Google Scholar]
- Dalpozzo, R. Catalytic asymmetric synthesis of hetero-substituted oxindoles. Org. Chem. Front. 2017, 4, 2063–2078. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Recent advances in organocatalytic methods for the synthesis of disubstituted 2- and 3-indolinones. Chem. Soc. Rev. 2012, 41, 7247–7290. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Chimni, S.S.; Mahajana, S.; Kumarb, A. Stereoselective synthesis of 3-amino-2-oxindoles from isatin imines: New scaffolds for bioactivity evaluation. RSC Adv. 2015, 5, 52481–52496. [Google Scholar] [CrossRef]
- Mohammadi, S.; Heiran, R.; Herrera, R.P.; Marqués-López, E. Isatin as a strategic motif for asymmetric catalysis. ChemCatChem 2013, 5, 2131–2148. [Google Scholar] [CrossRef]
- Vilaivan, T.; Bhanthumnavin, W. Organocatalyzed asymmetric α-oxidation, α-aminoxylation and α-amination of carbonyl compounds. Molecules 2010, 15, 917–958. [Google Scholar] [CrossRef] [PubMed]
- Vallribera, A.; Sebastian, R.M.; Shafir, A. Azodicarboxylates as electrophilic aminating reagents. Curr. Org. Chem. 2011, 15, 1539–1577. [Google Scholar] [CrossRef]
- Russo, A.; De Fusco, C.; Lattanzi, A. Enantioselective organocatalytic α-heterofunctionalization of active methines. RSC Adv. 2012, 2, 385–397. [Google Scholar] [CrossRef]
- Govender, T.; Arvidsson, P.I.; Maguire, G.E.M.; Kruger, H.G.; Naicker, T. Enantioselective organocatalyzed Transformations of β-ketoesters. Chem. Rev. 2016, 116, 9375–9437. [Google Scholar] [CrossRef] [PubMed]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Chimni, S.S. Organocatalytic asymmetric synthesis of 3-amino-2-oxindole derivatives bearing a tetra-substituted stereocenter. Tetrahedron Asymmetry 2013, 24, 343–356. [Google Scholar] [CrossRef]
- Yu, J.S.; Zhou, F.; Liu, Y.L.; Zhou, J.A. Journey in the catalytic synthesis of 3-substituted 3-aminooxindoles. Synlett 2015, 26, 2491–2504. [Google Scholar]
- Freckleton, M.; Baeza, A.; Benavent, L.; Chinchilla, R. Asymmetric organocatalytic electrophilic heterofunctionalization of oxindoles. Asian J. Org. Chem. 2018. [Google Scholar] [CrossRef]
- Almaşi, D.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Chiral 2-aminobenzimidazoles as recoverable organocatalysts for the addition of 1,3-dicarbonyl compounds to nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Conjugate addition of 1,3-dicarbonyl compounds to maleimides using a chiral C2-symmetric bis(2-aminobenzimidazole) as recyclable organocatalyst. Org. Lett. 2011, 13, 6106–6109. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Enantioselective synthesis of succinimides by michael addition of 1,3-dicarbonyl compounds to maleimides catalyzed by a chiral bis(2-aminobenzimidazole) organocatalyst. Eur. J. Org. Chem. 2013, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Trillo, P.; Baeza, A.; Nájera, C. Bis(2-aminobenzimidazole)-organocatalyzed asymmetric alkylation of activated methylene compounds with benzylic and allylic alcohols. Synthesis 2014, 46, 3399–3407. [Google Scholar]
- Serrano-Sánchez, D.; Baeza, A.; Alonso, D.A. Organocatalytic asymmetric α-chlorination of 1,3-dicarbonyl compounds catalyzed by 2-aminobenzimidazole derivatives. Symmetry 2016, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Trillo, P.; Gómez-Martínez, M.; Alonso, D.A.; Baeza, A. 2-aminobenzimidazole organocatalyzed asymmetric amination of cyclic 1,3-dicarbonyl compounds. Synlett 2015, 26, 95–100. [Google Scholar]
- Benavent, L.; Puccetti, F.; Baeza, A.; Gómez-Martínez, M. Readily available chiral benzimidazoles-derived guanidines as organocatalysts in the asymmetric α-amination of 1,3-dicarbonyl compounds. Molecules 2017, 22, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Qian, ZQ.; Zhou, F.; Du, T.P.; Wang, B.L.; Ding, M.; Zhao, X.L.; Zhou, J. Asymmetric construction of quaternary stereocenters by direct organocatalytic amination of 3-substituted oxindoles. Chem. Commun. 2009, 6753–6755. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Liu, L.; Wang, D.; Chen, Y.J. Highly enantioselective and organocatalytic α-amination of 2-oxindoles. Org. Lett. 2009, 11, 3874–3877. [Google Scholar] [PubMed]
- Zhou, F.; Ding, M.; Liu, Y.L.; Wang, C.H.; Ji, C.B.; Zhang, Y.Y.; Zhou, J. Organocatalytic asymmetric α-amination of unprotected 3-aryl and 3-aliphatic substituted oxindoles using di-tert-butyl azodicarboxylate. Adv. Synth. Catal. 2011, 353, 2945–2952. [Google Scholar] [CrossRef]
- Mouri, S.; Chen, Z.; Mitsunuma, H.; Furutachi, M.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric synthesis of 3-aminooxindoles: Enantiofacial selectivity switch in bimetallic vs monometallic schiff base catalysis J. Am. Chem. Soc. 2010, 132, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3aa–3ia are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. (°C) | Conv. (%) b | ee (%) c |
| 1 | 4 | PhMe | 25 | 90 | 13 |
| 2 | 5 | PhMe | 25 | 80 | 73 |
| 3 | 6 | PhMe | 25 | 80 | 57 |
| 4 | 7 | PhMe | 25 | 92 | 5 |
| 5 | 8 | PhMe | 25 | 40 | rac. |
| 6 | 9 | PhMe | 25 | 87 | 3 |
| 7 | 5 | CH2Cl2 | 25 | 80 | 56 |
| 8 | 5 | Et2O | 25 | 85 | 66 |
| 9 | 5 | THF | 25 | 75 | 50 |
| 10 | 5 | C6H14 | 25 | 81 | 73 |
| 11 | 5 | MeOH | 25 | 79 | 5 |
| 12 | 5 | PhMe | 0 d | 89 | 81 |
| 13 | 5 | C6H14 | 0 d | 82 | 51 |
| 14 | 5 | PhMe | −20 d | 56 | 71 |
| 15 | 5 | PhMe | −78 e | 59 | 70 |
 | ||||
|---|---|---|---|---|
| Entry | 1 | 3 | Yield (%) b | ee (%) c |
| 1 | 1a (R1 = Me, R2 = H) |  | 78 | 81 |
| 2 | 1b (R1 = Me, R2 = Br) |  | 84 | 80 |
| 3 | 1c (R1 = Ph, R2 = H) |  | 61 | 70 |
| 4 | 1d (R1 = Allyl, R2 = H) |  | 87 | 84 |
| 5 | 1e (R1 = CH2CO2Et, R2 = H) |  | 71 | 85 |
| 6 | 1f (R1 = Bn, R2 = H) |  | 83 | 70 |
| 7 | 1g (R1 = p-MeOBn, R2 = H) |  | 75 | 93 |
| 8 | 1h (R1 = p-NO2Bn, R2 = H) |  | 79 | 80 |
| 9 | 1i (R1 = p-CF3Bn, R2 = H) |  | 76 | 99 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benavent, L.; Baeza, A.; Freckleton, M. Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles. Molecules 2018, 23, 1374. https://doi.org/10.3390/molecules23061374
Benavent L, Baeza A, Freckleton M. Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles. Molecules. 2018; 23(6):1374. https://doi.org/10.3390/molecules23061374
Chicago/Turabian StyleBenavent, Llorenç, Alejandro Baeza, and Megan Freckleton. 2018. "Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles" Molecules 23, no. 6: 1374. https://doi.org/10.3390/molecules23061374
APA StyleBenavent, L., Baeza, A., & Freckleton, M. (2018). Chiral 2-Aminobenzimidazole as Bifunctional Catalyst in the Asymmetric Electrophilic Amination of Unprotected 3-Substituted Oxindoles. Molecules, 23(6), 1374. https://doi.org/10.3390/molecules23061374