Isolation, Structural Elucidation, and α-Glucosidase Inhibitory Activities of Triterpenoid Lactones and Their Relevant Biogenetic Constituents from Ganoderma resinaceum


 ,
, 
Abstract
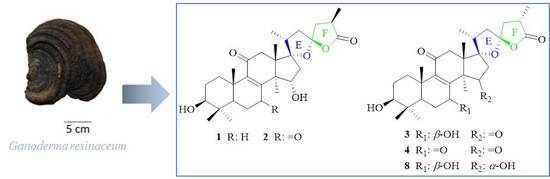
:
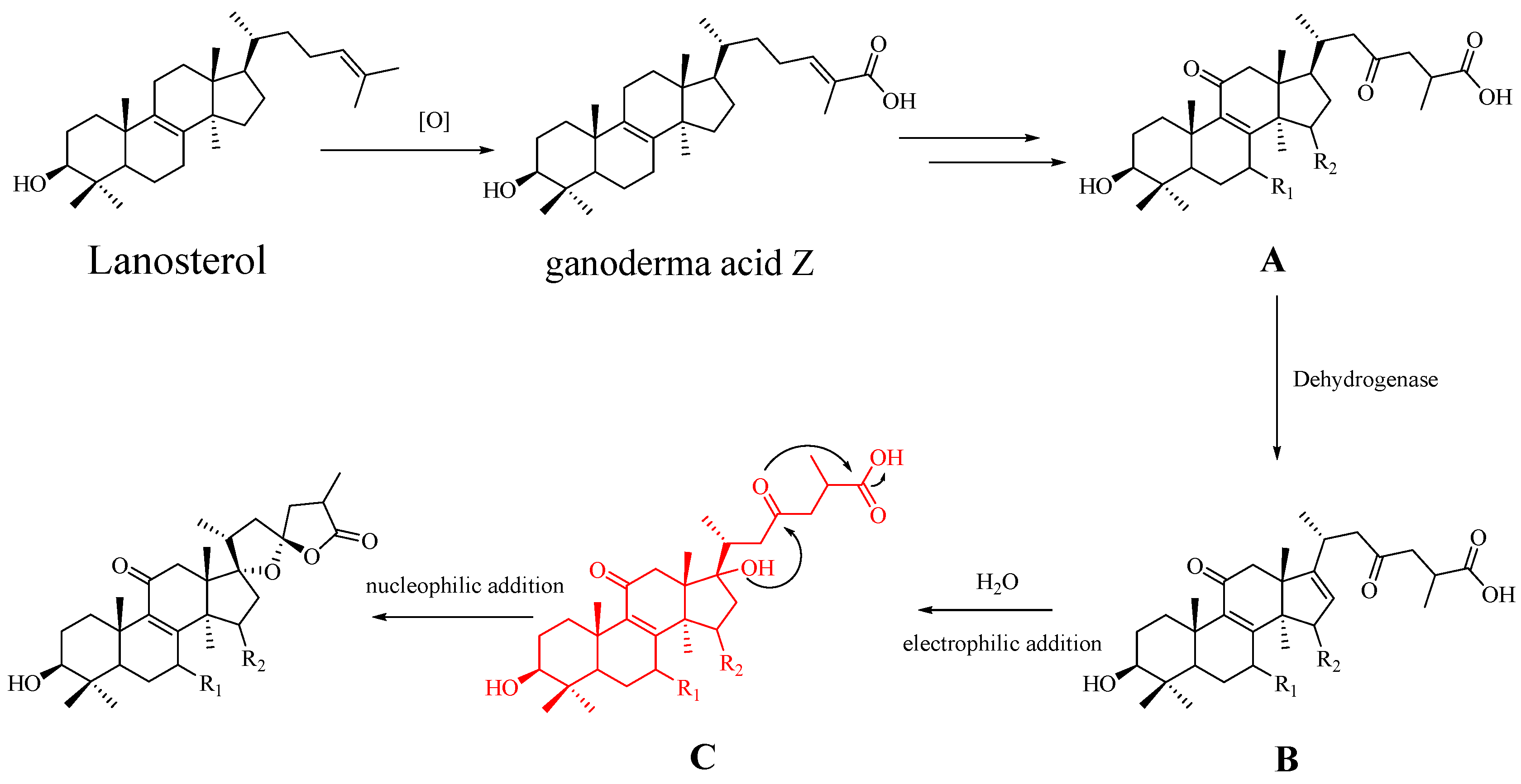
1. Introduction
2. Results
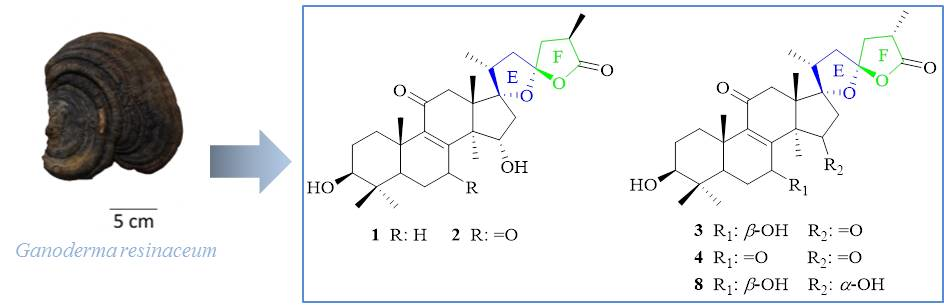
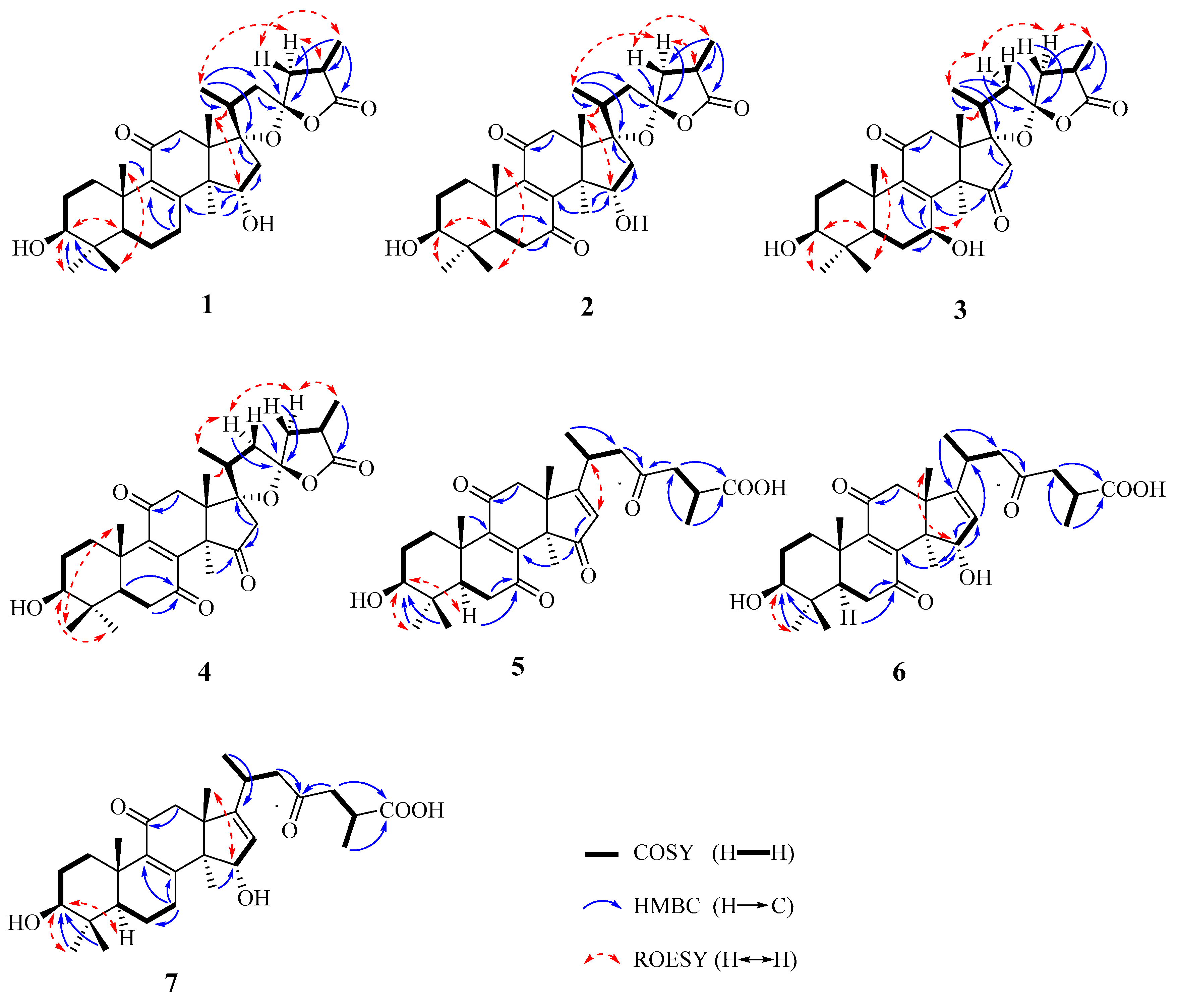
2.1. Structural Elucidation
2.2. Bioassay
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Fungal Materials
4.3. Extraction and Isolation
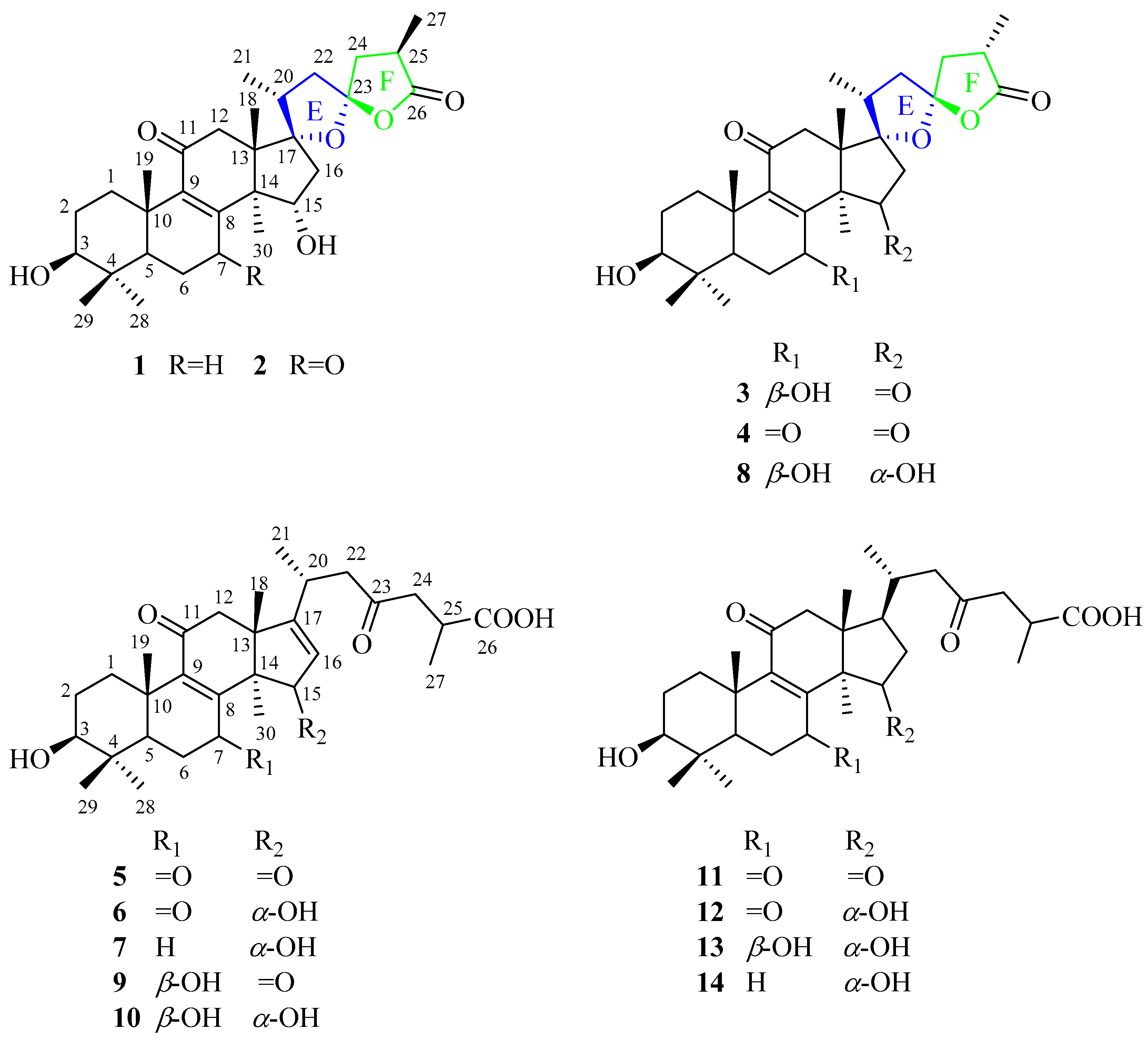
4.3.1. (17S,23S)-17,23-Epoxy-3β,15α-dihydroxy-11-oxo-5α-lanosta-8-en-26,23-olide (1)
4.3.2. (17S,23S)-17,23-Epoxy-3β,15α-dihydroxy-7,11-dioxo-5α-lanosta-8-en-26,23-olide (2)
4.3.3. (17S,23S)-17,23-Epoxy-3β,7β-dihydroxy-11,15-dioxo-5α-lanosta-8-en-26,23-olide (3)
4.3.4. (17S,23S)-17,23-Epoxy-3β,7β,15α-trihydroxy-11-oxo-5α-lanosta-8-en-26,23-olide (4)
4.3.5. 3β-Hydroxy-7,11,15,23-tetraoxo-5α-lanosta-8,16-dien-26-oic acid (5)
4.3.6. 3β,15α-Dihydroxy-7,11,23-trioxo-5α-lanosta-8,16-dien-26-oic acid (6)
4.3.7. 3β,15α-Dihydroxy-11,23-dioxo-5α-lanosta-8,16-dien-26-oic acid (7)
4.3.8. Bioassay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oyetayo, O.V. Medicinal Uses of Mushrooms in Nigeria: Towards Full and Sustainable Exploitation. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-R.; Liu, J.-Q.; Han, Z.-H.; Yuan, X.-X.; Luo, H.-R.; Qiu, M.-H. Protective effects of triterpenoids from Ganoderma resinaceum on H2O2-induced toxicity in HepG2 cells. Food Chem. 2013, 141, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Q.; Zhao, J.; Chen, L.-X.; Wang, S.-F.; Wang, Y.; Li, S.-P. Lanostane triterpenes from the mushroom Ganoderma resinaceum and their inhibitory activities against α-glucosidase. Phytochemistry 2018, 149, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Chen, L.X.; Zhao, J.; Tang, P.; Li, S.P. Nortriterpenoids from the Fruiting Bodies of the Mushroom Ganoderma resinaceum. Molecules 2017, 22, 1073. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Chen, L.X.; Li, S.P.; Zhao, J. Meroterpenoids from the fruiting bodies of higher fungus Ganoderma resinaceum. Phytochem. Lett. 2017, 22, 214–218. [Google Scholar] [CrossRef]
- Chen, X.Q.; Chen, L.X.; Li, S.P.; Zhao, J. A new nortriterpenoid and an ergostane-type steroid from the fruiting bodies of the fungus Ganoderma resinaceum. J. Asian Nat. Prod. Res. 2017, 19, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Zengin, G.; Sarikurkcu, C.; Gunes, E.; Uysal, A.; Ceylan, R.; Uysal, S.; Gungor, H.; Aktumsek, A. Two Ganoderma species: Profiling of phenolic compounds by HPLC-DAD, antioxidant, antimicrobial and inhibitory activities on key enzymes linked to diabetes mellitus, Alzheimer’s disease and skin disorders. Food Funct. 2015, 6, 2794–2802. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.E.; Carbonero, E.R.; Simao, R.D.C.G.; Kadowaki, M.K.; Sassaki, G.L.; Osaku, C.A.; Gorin, P.A.J.; Iacomini, M. An unusual water-soluble beta-glucan from the basidiocarp of the fungus Ganoderma resinaceum. Carbohydr. Polym. 2008, 72, 473–478. [Google Scholar] [CrossRef]
- Niu, X.-M.; Li, S.-H.; Xiao, W.-L.; Sun, H.-D.; Che, C.-T. Two new lanostanoids from Ganoderma resinaceum. J. Asian Nat. Prod. Res. 2007, 9, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Miranda, A.; Fernandes, E.; Santos, S.; Fraga, I.; Santos, D.L.; Dias, A.A.; Bezerra, R.M. Endopolysaccharides from Ganoderma resinaceum, Phlebia rufa, and Trametes versicolor Affect Differently the Proliferation Rate of HepG2 Cells. Appl. Biochem. Biotechnol. 2013, 169, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Ofodile, L.N.; Uma, N.U.; Kokubun, T.; Grayer, R.J.; Ogundipe, O.T.; Simmonds, M.S.J. Antimicrobial activity of some Ganoderma species from Nigeria. Phytother. Res. 2005, 19, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Gao, Y.-X.; Jin, H.-Z.; Shan, L.; Chang, W.-L.; Yang, X.-W.; Zeng, H.-W.; Wang, N.; Steinmetz, A.; Zhang, W.-D. Chemical constituents of Abies fabri. Phytochemistry 2015, 117, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W.; Lv, C.; Fang, X.; Tian, X.-H.; Ye, J.; Li, H.-L.; Shan, L.; Shen, Y.-H.; Zhang, W.-D. Eight Pairs of Epimeric Triterpenoids Involving a Characteristic Spiro-E/F Ring from Abies faxoniana. J. Nat. Prod. 2015, 78, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-Y.; Chen, H.; Liu, C.; Wang, H.-Q.; Kang, J.; Li, Y.; Chen, R.-Y. Triterpenoids of Ganoderma theaecolum and their hepatoprotective activities. Fitoterapia 2014, 98, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-S.; Wang, Y.-G.; Ma, Q.-Y.; Huang, S.-Z.; Hu, L.-L.; Dai, H.-F.; Yu, Z.-F.; Zhao, Y.-X. Three New Lanostanoids from the Mushroom Ganoderma tropicum. Molecules 2015, 20, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.-H.; Yang, M.; Wang, X.-M.; Xia, J.-M.; Zhang, Z.-M.; Liu, X.; Guo, D.-A. Spectral assignments and reference data structure elucidation and complete NMR spectral assignments of three new lanostanoid triterpenes with unprecedented Δ16,17 double bond from Ganoderma lucidum. Magn. Reson. Chem. 2007, 45, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.-L.; Ma, Q.-Y.; Huang, S.-Z.; Guo, Z.-K.; Ma, H.-X.; Guo, J.-C.; Dai, H.-F.; Zhao, Y.-X. Three new lanostanoid triterpenes from the fruiting bodies of Ganoderma tropicum. J. Asian Nat. Prod. Res. 2013, 15, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Kuo, S.H.; Won, S.J. Steroids of Formosan Ganoderma amboinense. Phytochemistry 1993, 32, 1549–1551. [Google Scholar]
- Kikuchi, T.; Kanomi, S.; Kadota, S.; Murai, Y.; Tsubono, K.; Ogita, Z.I. Constituents of the fungus Ganoderma lucidum (Fr.) Karst. I. Structures of ganoderic acids C2, E, I, and K, lucidenic acid F and related compounds. Chem. Pharm. Bull. 1986, 34, 3695–3712. [Google Scholar] [CrossRef]
- Kikuchi, T.; Kanomi, S.; Murai, Y.; Kadota, S.; Tsubono, K.; Ogita, Z. Constituents of the fungus Ganoderma lucidum (Fr.) Karst. III. Structures of ganolucidic acids A and B, new lanostane-type triterpenoids. Chem. Pharm. Bull. 1986, 34, 4030–4036. [Google Scholar] [CrossRef]
- Hasegawa, S.; Kaneko, N.; Hirose, Y. Triterpenes from the seed of Abies firma. Phytochemistry 1987, 26, 1095–1099. [Google Scholar] [CrossRef]
- Li, D.-Q.; Qian, Z.-M.; Li, S.-P. Inhibition of Three Selected Beverage Extracts on α-Glucosidase and Rapid Identification of Their Active Compounds Using HPLC-DAD-MS/MS and Biochemical Detection. J. Agric. Food. Chem. 2010, 58, 6608–6613. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–14 are available from the corresponding authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | 1 a | 2 | 3 | 4 | 8 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | δC | δH | |
| 1 | 35.21, CH2 | 1.36, dt (13.2, 3.0); 3.51 m | 34.1, CH2 | 1.24, m; 2.83, dt (13.2, 3.0) | 34.7, CH2 | 0.97 dd (13.2, 4.2); 2.83, dt (13.2, 3.6) | 33.7, CH2 | 1.34, dt (13.8, 3.6); 2.84, dt (13.8, 3.6) | 34.5, CH2 | 0.92, m; 2.75, dt (13.8, 3.6) |
| 2 | 29.0, CH2 | 1.99, m; 2.06, dt (13.8, 3.0) | 27.5, CH2 | 1.70, dd (12.0, 3.6); 1.76, m | 27.7, CH2 | 1.67, m | 27.3, CH2 | 1.68, dt (12.0, 3.6); 1.76, m | 27.5, CH2 | 1.65, m; 2.11, dd (12.6, 7.2) |
| 3 | 77.8, CH | 3.52, dd overlapped | 77.3, CH | 3.29, dd (11.4, 4.8) | 78.2, CH | 3.22, dd (11.4, 4.8) | 77.5, CH | 3.30, dd (11.4, 4.2) | 78.2, CH | 3.23, dd (11.4, 4.8) |
| 4 | 39.7, C | 38.7, C | 38.9, C | 39.0, C | 38.6, C | |||||
| 5 | 52.4, CH | 1.18, br d (12.0) | 49.3, CH | 1.55, dd (9.6, 7.8) | 49.0, CH | 0.88, br d (12.0) | 49.9, CH | 1.63, dd overlapped | 48.9, CH | 0.92, d (12.0) |
| 6 | 17.9, CH2 | 1.51, m; 1.84, m | 36.2, CH2 | 2.56, m; 2.57, m | 26.6, CH2 | 1.59, dt (13.2, 3.6); 2.19, dd (13.2, 8.4) | 35.9, CH2 | 2.58, m; 2.62, m | 27.7, CH2 | 1.61, m; 1.65, m |
| 7 | 30.7, CH2 | 2.76, dd (21.0, 5.4); 2.96, m | 205.0, C | 66.7, CH | 4.81, m | 198.5, C | 69.0, CH | 4.60, br t (6.6) | ||
| 8 | 165.2, C | 150.3, C | 157.5, C | 148.9, C | 158.7, C | |||||
| 9 | 139.9, C | 154.5, C | 142.4, C | 151.5, C | 141.6, C | |||||
| 10 | 38.4, C | 40.0, C | 38.6, C | 40.4, C | 38.5, C | |||||
| 11 | 198.6, C | 201.8, C | 198.1, C | 200.2, C | 200.2, C | |||||
| 12 | 47.4, CH2 | 2.46, d (16.8); 3.38, d (16.8) | 47.2, CH2 | 2.35, d (16.8); 3.25, d (16.8) | 44.1, CH2 | 2.48, d (16.8); 3.17, d (16.8) | 44.7, CH2 | 2.48, d (16.2); 3.20, d (16.2) | 47.0, CH2 | 2.27, d (15.0); 3.12, d (15.0) |
| 13 | 50.2, C | 50.7 C | 48.1, C | 47.3, C | 49.5, C | |||||
| 14 | 54.4, C | 53.5, C | 59.1, C | 56.5, C | 54.3, C | |||||
| 15 | 72.9, CH | 4.72, dd (9.0, 7.2) | 72.7, CH | 4.45, dd (9.0, 7.2) | 216.1, C | 207.1, C | 73.0, CH | 4.80, t (8.4) | ||
| 16 | 47.3, CH2 | 2.52, dd (15.0, 9.0); 2.80, dd (15.0, 7.2) | 44.3, CH2 | 2.32, dd (15.6, 9.0); 2.53, dd (15.6, 7.2) | 48.0, CH2 | 2.47, d (20.4); 3.35, d (20.4) | 48.1, CH2 | 2.45, d (16.2); 3.33, d (16.2) | 44.7, CH2 | 2.29, m; 2.41, dd (15.0, 8.4) |
| 17 | 95.6, C | 94.7, C | 91.8, C | 92.2, C | 94.7, C | |||||
| 18 | 19.8, CH3 | 1.15, s | 20.0, CH3 | 1.05, s | 20.9, CH3 | 1.18, s | 20.0, CH3 | 1.03, s | 19.7, CH3 | 1.16, s |
| 19 | 19.4, CH3 | 1.42, s | 17.4, CH3 | 1.29, s | 18.3, CH3 | 1.21, s | 17.7, CH3 | 1.20, s | 19.4, CH3 | 1.26, s |
| 20 | 43.7, CH | 2.14, m | 43.6, CH | 2.26, m | 43.1, CH | 2.38, m | 43.3, CH | 2.38, m | 48.3, CH | 2.29, m |
| 21 | 17.9, CH3 | 0.91, d (6.6) | 18.1, CH3 | 1.01, d (6.6) | 18.2, CH3 | 1.12, d (6.6) | 18.3, CH3 | 1.10, d (7.2) | 18.1, CH3 | 1.02, d (7.2) |
| 22 | 44.61, CH2 | 1.76 d (13.8); 2.64, dd (13.8, 6.6) | 44.6, CH2 | 1.81, d (13.8); 2.73, dd (13.8, 6.6) | 44.4, CH2 | 1.93, d (14.4); 2.73, dd (14.4, 7.2) | 44.3, CH2 | 1.91, d (14.4); 2.73, dd (14.4, 6.6) | 44.6, CH2 | 1.84, d (14.4); 2.72, dd (14.4, 6.6) |
| 23 | 113.3, C | 112.7, C | 113.0, C | 112.9, C | 113.3, C | |||||
| 24 | 44.62, CH2 | 1.96, d (12.6); 2.36, dd (12.6, 8.4) | 44.9, CH2 | 2.03, dd (12.6, 11.4); 2.47, dd (12.6, 8.4) | 44.5, CH2 | 2.10, dd (13.2, 11.4); 2.57, dd (13.2, 8.4) | 44.6, CH2 | 2.09, dd (12.6, 11.4); 2.56, dd overlapped | 44.7, CH2 | 2.05, dd (12.6, 11.4); 2.49, dd (12.6, 8.4) |
| 25 | 35.7, CH | 3.02, m | 35.4, CH | 2.92, m | 35.4, CH | 2.92, m | 35.3, CH | 2.94, m | 35.5, CH | 2.96, m |
| 26 | 178.7, C | 178.5, C | 178.3, C | 178.2, C | 179.2, C | |||||
| 27 | 15.1, CH3 | 1.23, d (7.2) | 14.9, CH3 | 1.25, d (6.6) | 14.9, CH3 | 1.29, d (7.2) | 14.9, CH3 | 1.28, d (7.2) | 14.9, CH3 | 1.28, d (7.2) |
| 28 | 28.9, CH3 | 1.27, s | 27.7, CH3 | 1.03, s | 28.2, CH3 | 1.04, s | 27.7, CH3 | 1.03, s | 28.1, CH3 | 1.03, s |
| 29 | 16.8, CH3 | 1.13, s | 15.4, CH3 | 0.89, s | 15.5, CH3 | 0.87, s | 15.4, CH3 | 0.89, s | 15.7, CH3 | 0.85, s |
| 30 | 21.8, CH3 | 1.68, s | 22.6, CH3 | 1.24, s | 26.9, CH3 | 1.41, s | 25.8, CH3 | 1.54, s | 21.2, CH3 | 1.33, s |
| No | 5 | 6 | 7 | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 1 | 35.2, CH2 | 1.37, dd (13.2, 4.2); 2.85, m | 35.4, CH2 | 1.29, m; 2.92, dt (13.8, 3.6) | 35.8, CH2 | 1.08, dd (13.2, 3.6); 2.99, dt (13.2, 3.6) |
| 2 | 28.1, CH2 | 1.62, m | 28.6, CH2 | 1.69, m; 1.74, m | 28.6, CH2 | 1.62, m; 1.68, m |
| 3 | 78.4, CH | 3.18, dd (11.4, 4.8) | 78.2, CH | 3.22, dd (11.4, 4.8) | 79.5, CH | 3.17, dd (11.4, 4.8) |
| 4 | 40.1, C | 40.1, C | 40.3, C | |||
| 5 | 50.5, CH | 1.71, dd (13.8, 4.2) | 51.5, CH | 1.58, dd (15.0, 1.8) | 53.7, CH | 0.95, d (13.2) |
| 6 | 36.0, CH2 | 2.49, d (13.8); 2.55, dd (13.8, 4.2) | 36.8, CH2 | 2.55, m; 2.70, t (15.0) | 18.7, CH2 | 1.51, m; 1.81, m |
| 7 | 200.3, C | 206.3, C | 32.9, CH2 | 2.55, m | ||
| 8 | 151.5, C | 150.9, C | 166.5, C | |||
| 9 | 154.0, C | 155.94, C | 140.9, C | |||
| 10 | 42.0, C | 41.8, C | 39.4, C | |||
| 11 | 200.8, C | 202.6, C | 201.2, C | |||
| 12 | 45.6, CH2 | 2.54, d (16.8); 3.09, d (16.8) | 48.9, CH2 | 2.42, d (16.2); 3.08, d (16.2) | 48.7, CH2 | 2.24, d (16.8); 2.94, d (16.8) |
| 13 | 53.2, C | 53.8, C | 53.4, C | |||
| 14 | 56.6, C | 55.3, C | 58.0, C | |||
| 15 | 206.7, C | 78.4, CH | 4.95, br s | 77.8, CH | 4.90, br s | |
| 16 | 124.0, CH | 5.65, s | 124.5, CH | 5.26, s | 125.7, CH | 5.18, s |
| 17 | 186.7, C | 155.98, C | 156.3, C | |||
| 18 | 30.1, CH3 | 1.02, s | 23.3, CH3 | 0.95, s | 23.0, CH3 | 0.96, s |
| 19 | 17.7, CH3 | 1.08, s | 17.7, CH3 | 1.30, s | 19.4, CH3 | 1.12, s |
| 20 | 30.7, CH | 2.93, m | 28.5, CH | 2.63, m | 28.9, CH | 2.62, m |
| 21 | 20.1, CH3 | 1.05, d (6.6) | 21.0, CH3 | 1.04, d (6.6) | 21.0, CH3 | 1.04, d (7.2) |
| 22 | 48.5, CH2 | 2.70, dd (17.4, 5.4); 2.88, d (17.4) | 49.4, CH2 | 2.57, d (16.8); 2.77, dd (16.8, 7.2) | 49.5, CH2 | 2.54, d (16.8); 2.73, dd (16.8, 7.2) |
| 23 | 209.0, C | 209.9, C | 210.0, C | |||
| 24 | 47.3, CH2 | 2.49, m; 2.81, m | 47.5, CH2 | 2.53, m; 2.85, m | 47.5, CH2 | 2.52, dd (13.2, 8.4); 2.85, dd (13.2, 9.0) |
| 25 | 36.2, CH | 2.78, m | 36.0, CH | 2.82, m | 36.1, CH | 2.83, m |
| 26 | 179.6, C | 179.9, C | 179.8, C | |||
| 27 | 17.7, CH3 | 1.09, d (7.2) | 17.7, CH3 | 1.14, d (7.2) | 17.7, CH3 | 1.15, d (6.6) |
| 28 | 28.3, CH3 | 0.96, s | 28.3, CH3 | 1.00, s | 29.1, CH3 | 1.02, s |
| 29 | 16.0, CH3 | 0.83, s | 16.2, CH3 | 0.89, s | 16.6, CH3 | 0.82, s |
| 30 | 33.6, CH3 | 1.39, s | 23.2, CH3 | 1.17, s | 22.7, CH3 | 1.21, s |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-Q.; Lin, L.-G.; Zhao, J.; Chen, L.-X.; Tang, Y.-P.; Luo, D.-L.; Li, S.-P. Isolation, Structural Elucidation, and α-Glucosidase Inhibitory Activities of Triterpenoid Lactones and Their Relevant Biogenetic Constituents from Ganoderma resinaceum. Molecules 2018, 23, 1391. https://doi.org/10.3390/molecules23061391
Chen X-Q, Lin L-G, Zhao J, Chen L-X, Tang Y-P, Luo D-L, Li S-P. Isolation, Structural Elucidation, and α-Glucosidase Inhibitory Activities of Triterpenoid Lactones and Their Relevant Biogenetic Constituents from Ganoderma resinaceum. Molecules. 2018; 23(6):1391. https://doi.org/10.3390/molecules23061391
Chicago/Turabian StyleChen, Xian-Qiang, Li-Gen Lin, Jing Zhao, Ling-Xiao Chen, Yu-Ping Tang, De-Lun Luo, and Shao-Ping Li. 2018. "Isolation, Structural Elucidation, and α-Glucosidase Inhibitory Activities of Triterpenoid Lactones and Their Relevant Biogenetic Constituents from Ganoderma resinaceum" Molecules 23, no. 6: 1391. https://doi.org/10.3390/molecules23061391
APA StyleChen, X. -Q., Lin, L. -G., Zhao, J., Chen, L. -X., Tang, Y. -P., Luo, D. -L., & Li, S. -P. (2018). Isolation, Structural Elucidation, and α-Glucosidase Inhibitory Activities of Triterpenoid Lactones and Their Relevant Biogenetic Constituents from Ganoderma resinaceum. Molecules, 23(6), 1391. https://doi.org/10.3390/molecules23061391