
Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation

 ,
,  and
and
Abstract
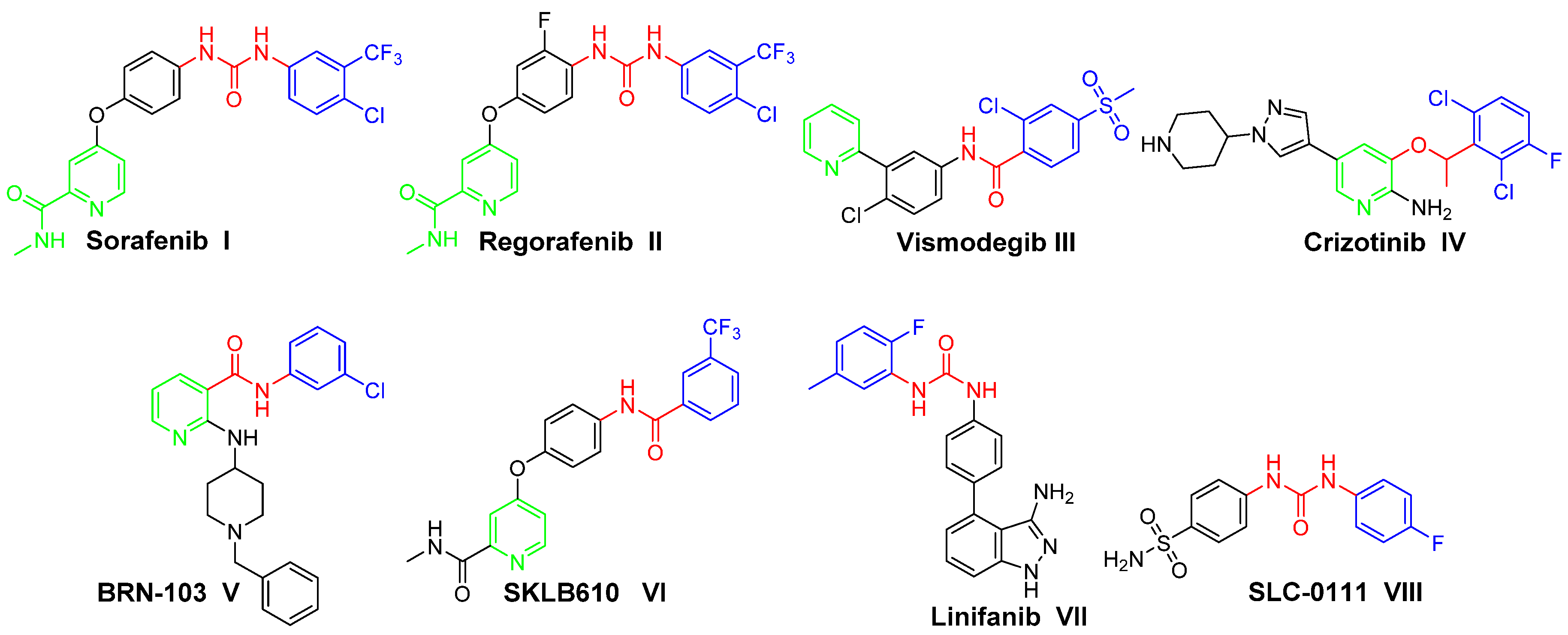
:1. Introduction
2. Results
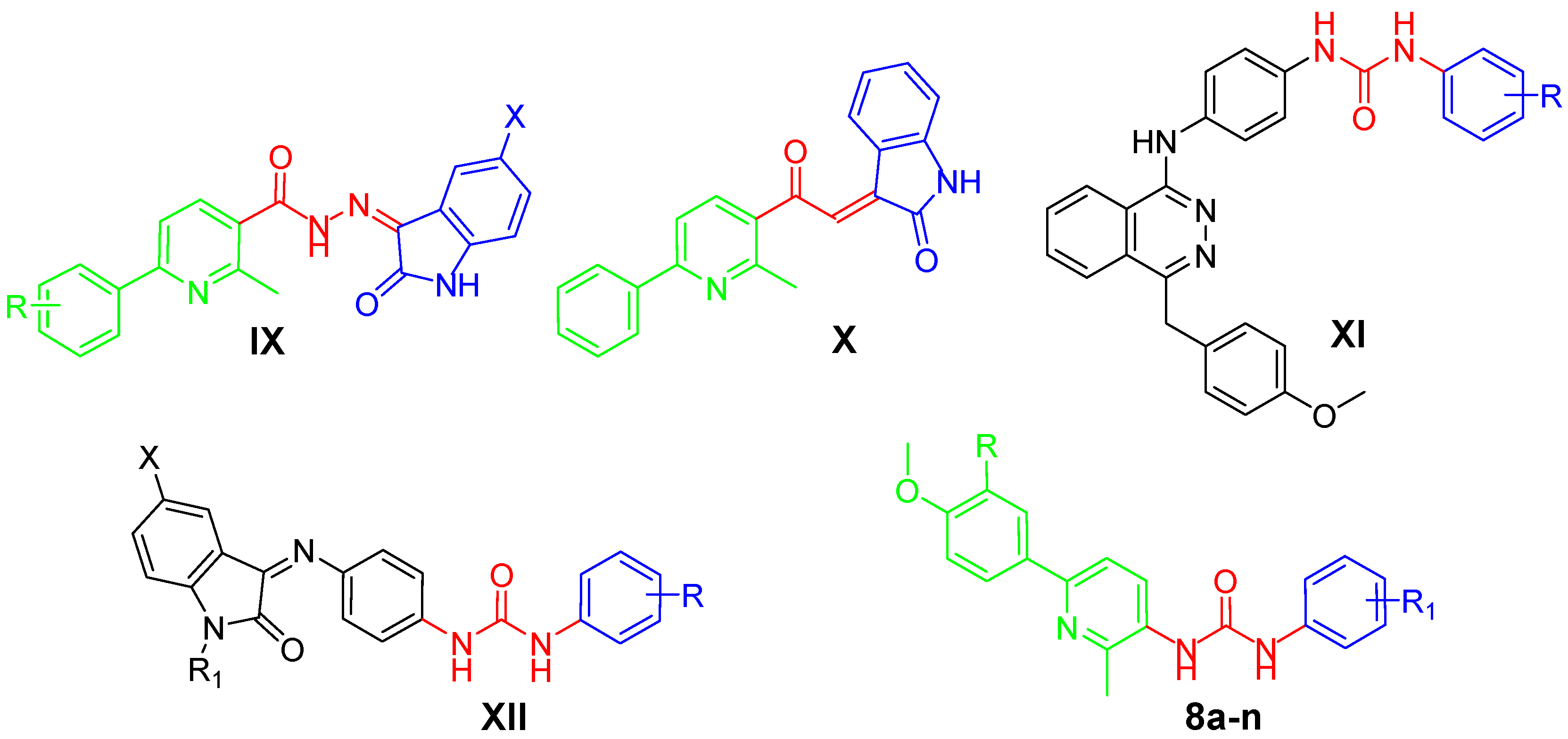
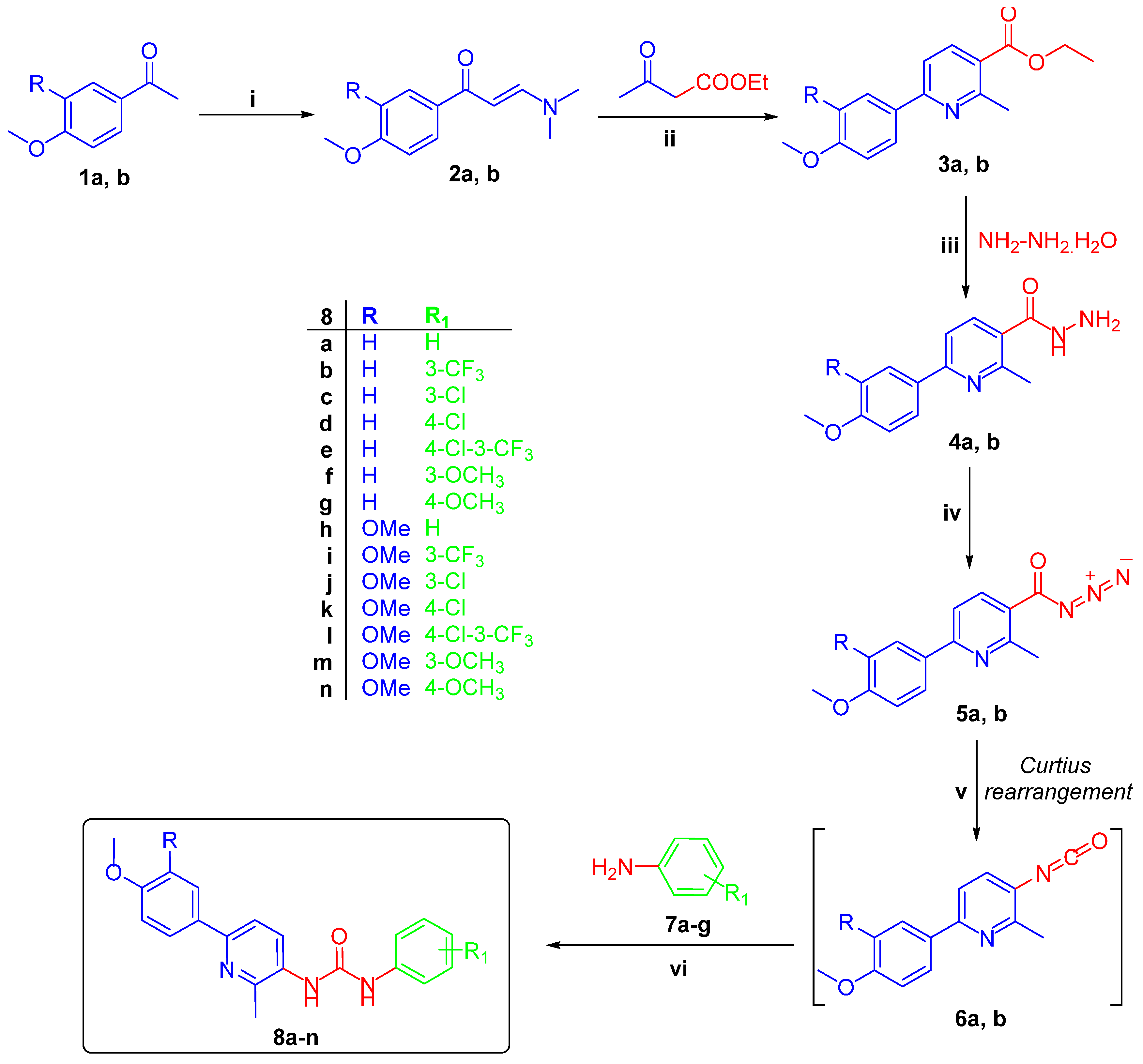
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Anti-Proliferative Activity
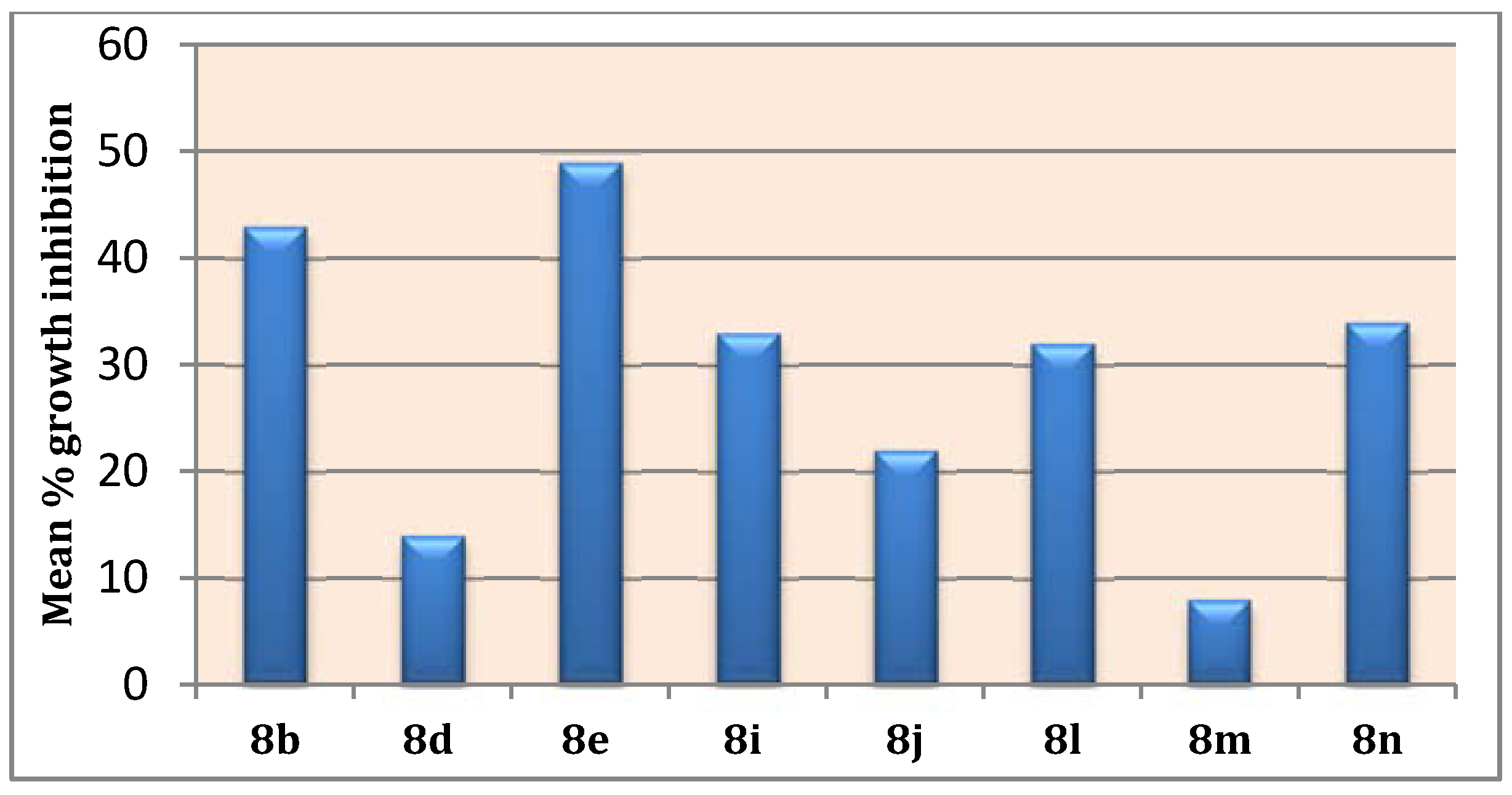
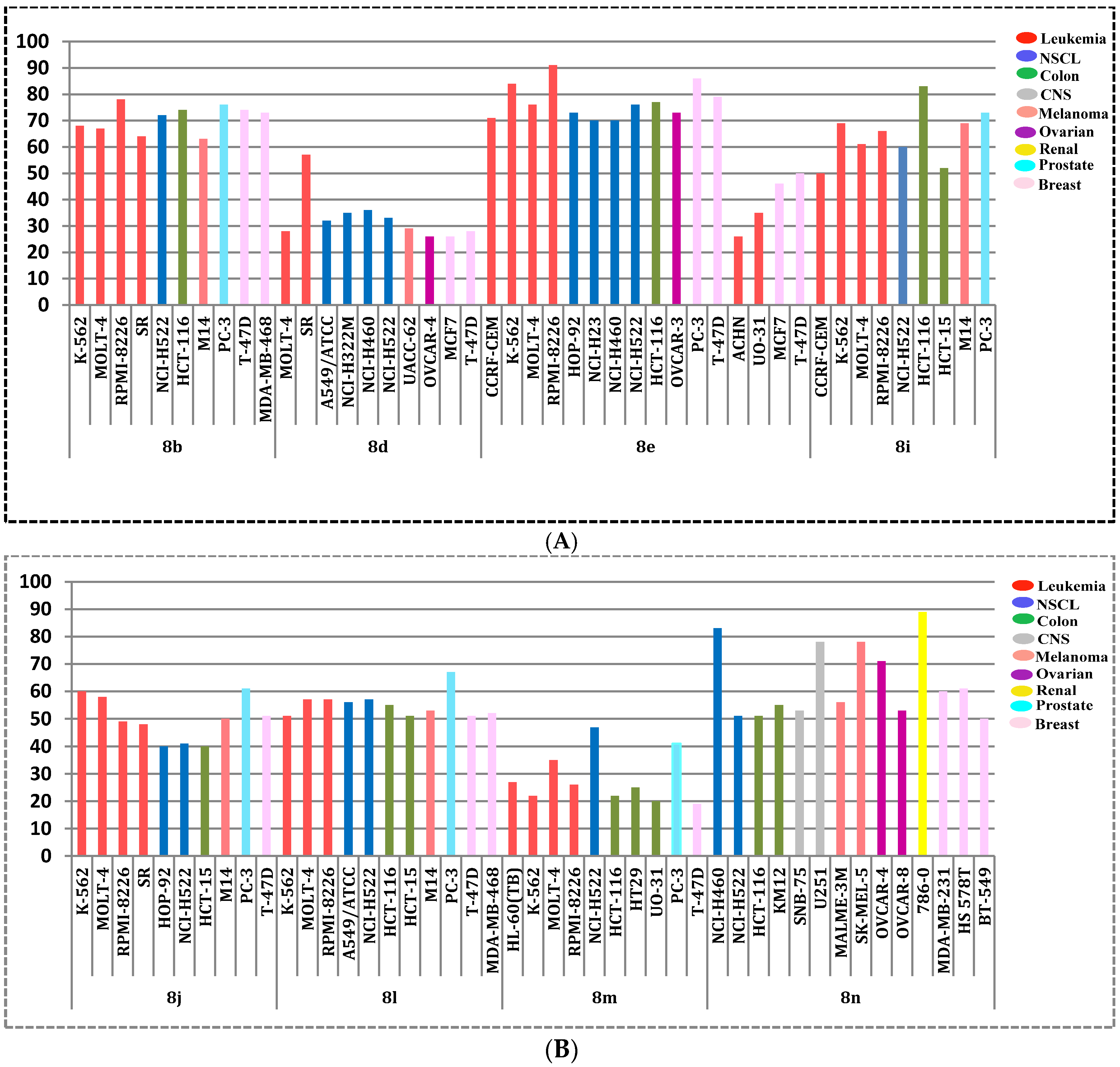
2.2.2. NCI, USA Cytotoxicity Assay towards 60 Cancer Cell Lines
2.2.3. Cytotoxic Activity against Non-Tumorigenic Human WI-38 Cells
2.2.4. VEGFR-2 Inhibitory Assay
2.3. Physicochemical Properties and ADME Profiling
3. Experimental
3.1. Chemistry
3.1.1. Synthesis of 6-(4-Methoxyphenyl/3,4-dimethoxyphenyl)-2-methylnicotinohydrazide 4a,b
3.1.2. General Procedure for the Preparation of Target Pyridine-Ureas 8a–n
3.2. Biological Evaluation
3.2.1. In Vitro Anti-Proliferative Activity towards Breast Cancer MCF7 Cell Line
3.2.2. In Vitro Cytotoxic Activity by NCI-USA
3.2.3. Measurement of Inhibitory Activity against VEGFR-2
3.2.4. Physicochemical Properties and ADME Profiling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Adami, H.; Hunter, D.; Trichopoulos, D. Textbook of Cancer Epidemiology; Oxford University Press: New York, NY, USA, 2002; pp. 301–373. [Google Scholar]
- International Agency for Research on Cancer. IGlobocan: Estimated Cancer Incidence, Mortality, and Prevalence Worldwide in 2012; IARC: Lyon, France, 2014. [Google Scholar]
- Davari, A.S.; Abnous, K.; Mehri, S.; Ghandadi, M.; Hadizadeh, F. Synthesis and biological evaluation of novel pyridine derivatives as potential anticancer agents and phosphodiesterase-3 inhibitors. Bioorg. Chem. 2014, 57, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Goh, A.W.; Yu, M.; Adams, J.; Lam, F.; Teo, T.; Li, P.; Noll, B.; Zhong, L.; Diab, S.; et al. Discovery of (E)-3-((Styrylsulfonyl)methyl)pyridine and (E)-2-((Styrylsulfonyl)methyl)pyridine Derivatives as Anticancer Agents: Synthesis, Structure–Activity Relationships, and Biological Activities. J. Med. Chem. 2014, 57, 2275–2291. [Google Scholar] [CrossRef] [PubMed]
- Helal, M.H.; El-Awdan, S.A.; Salem, M.A.; Abd-Elaziz, T.A.; Moahamed, Y.A.; El-Sherif, A.A.; Mohamed, G.A.M. Synthesis, biological evaluation and molecular modeling of novel series of pyridine derivatives as anticancer, anti-inflammatory and analgesic agents. Spectrochim. Acta Part A 2015, 135, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- DiGiulio, S. FDA Approves Stivarga for Advanced GIST. Oncol. Times 2013, 35, 12. [Google Scholar]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.E.; Yoo, M.S.; Choi, J.H.; Lee, J.Y.; Kim, J.H.; Kim, J.H.; Lee, J.K.; Kim, G.I.; Park, Y.; Chi, Y.H.; et al. BRN-103, a novel nicotinamide derivative, inhibits VEGF-induced angiogenesis and proliferation in human umbilical vein endothelial cells. Bioorg. Med. Chem. Lett. 2011, 21, 6236–6241. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.X.; Zheng, R.L.; Lin, H.J.; Luo, S.D.; Zhou, Y.; Xu, Y.Z.; Zeng, X.X.; Wang, Z.; Zhou, L.N.; Mao, Y.Q.; et al. SKLB610: A novel potential inhibitor of vascular endothelial growth factor receptor tyrosine kinases inhibits angiogenesis and tumor growth In Vivo. Cell. Physiol. Biochem. 2011, 27, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Lv, P.C.; Yan, T.; Zhu, H.L. Urea derivatives as anticancer agents. Anti Cancer Agents Med. Chem. 2009, 9, 471–480. [Google Scholar] [CrossRef]
- Tan, E.-H.; Goss, G.D.; Salgia, R.; Besse, B.; Gandara, D.R.; Hanna, N.H.; Yang, J.C.-H.; Thertulien, R.; Wertheim, M.; Mazieres, J. Phase 2 trial of Linifanib (ABT-869) in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Safety Study of SLC-0111 in Subjects with Advanced Solid Tumours. Available online: https://clinicaltrials.gov/ct2/show/NCT02215850 (accessed on 10 April 2018).
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Winum, J.-Y. Designing carbonic anhydrase inhibitors for the treatment of breast cancer. Expert Opin. Drug Discov. 2015, 10, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Altoukhy, A.; Mahrous, H.; Abdel-Aziz, H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015, 90, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Alsherbiny, M.A.; Aly, M.H.; Ghabbour, H.A.; Abdel-Aziz, H.A. Synthesis and cytotoxic activity of biphenylurea derivatives containing indolin-2-one moieties. Molecules 2016, 21, 762. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Aly, M.H.; Zada, S.; Ali, M.M.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; Abou El Ella, D.A. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, In Vitro biological evaluation and molecular docking. Eur. J. Med. Chem. 2015, 100, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Abou-Seri, S.M.; El Kerdawy, A.M.; Ayyad, R.R.; Hamdy, A.M.; Ghabbour, H.A.; Ali, M.M.; El Ella, D.A.A. Increasing the binding affinity of VEGFR-2 inhibitors by extending their hydrophobic interaction with the active site: Design, synthesis and biological evaluation of 1-substituted-4-(4-methoxybenzyl)phthalazine derivatives. Eur. J. Med. Chem. 2016, 113, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Fares, M.; Ceruso, M.; Ghabbour, H.A.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; El Ella, D.A.A.; Supuran, C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016, 110, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Ghabbour, H.; Al-Ansary, G.H.; Assaf, A.M.; Al-Dhfyan, A. Synthesis, crystal study, and anti-proliferative activity of some 2-benzimidazolylthioacetophenones towards triple-negative breast cancer MDA-MB-468 cells as apoptosis-inducing agents. Int. J. Mol. Sci. 2016, 17, 1221. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, H.A.; Ghabbour, H.A.; Eldehna, W.M.; Al-Rashood, S.T.; Al-Rashood, K.A.; Fun, H.K.; Al-Tahhan, M.; Al-Dhfyan, A. 2-((Benzimidazol-2-yl)thio)-1-arylethan-1-ones: Synthesis, crystal study and cancer stem cells CD133 targeting potential. Eur. J. Med. Chem. 2015, 104, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino)benzenesulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Al-Ansary, G.H.; Eldehna, W.M.; Ghabbour, H.A.; Al-Rashood, S.T.; Al-Rashood, K.A.; Eladwy, R.A.; Al-Dhfyan, A.; Kabil, M.M.; Abdel-Aziz, H.A. Cancer stem cells CD133 inhibition and cytotoxicity of certain 3-phenylthiazolo [3, 2-a] benzimidazoles: Design, direct synthesis, crystal study and In Vitro biological evaluation. J. Enzym. Inhib. Med. Chem. 2017, 32, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Boyd MRand Paull, K.D. Some practical considerations and applications of the National Cancer Institute In Vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Boyd, M.R. Cancer Drug Discovery and Development: Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials and Approval; Teicher, B.A., Ed.; Humana Press: Totowa, NJ, USA, 2014; Chapter 1; pp. 41–62. [Google Scholar]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Fares, M.; Abdel-Aziz, M.M.; Abdel-Aziz, H.A. Design, synthesis and antitubercular activity of certain nicotinic Acid hydrazides. Molecules 2015, 20, 8800–8815. [Google Scholar] [CrossRef] [PubMed]
- Almahli, H.; Hadchity, E.; Jaballah, M.Y.; Daher, R.; Ghabbour, H.A.; Kabil, M.M.; Al-shakliah, N.S.; Eldehna, W.M. Development of novel synthesized phthalazinone-based PARP-1 inhibitors with apoptosis inducing mechanism in lung cancer. Bioorg. Chem. 2018, 77, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; EL-Naggar, D.H.; Hamed, A.R.; Ibrahim, H.S.; Ghabbour, H.A.; Abdel-Aziz, H.A. One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells. J. Enzym. Inhib. Med. Chem. 2018, 33, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Abo-Ashour, M.F.; Ibrahim, H.S.; Al-Ansary, G.H.; Ghabour, H.A.; Elaasser, M.M.; Ahmed, H.Y.A.; Safwat, N.A. Novel [(3-indolylmethylene)hydrazono]indolin-2-ones as apoptotic anti-proliferativeagents: Design, synthesis and In Vitro biological evaluation. J. Enzym. Inhib. Med. Chem. 2018, 33, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; El Ella, D.A.A. 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and In Vitro biological evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 8a–n are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | R1 | IC50 (µM) a | |
|---|---|---|---|---|
| 48 h | 72 h | |||
| 8a | H | H | 3.03 ± 0.22 | 2.83 ± 0.25 |
| 8b | H | 3-CF3 | 5.66 ± 0.45 | 5.03 ± 0.29 |
| 8c | H | 3-Cl | 6.40 ± 0.41 | 3.52 ± 0.11 |
| 8d | H | 4-Cl | 7.10 ± 0.38 | 5.14 ± 0.46 |
| 8e | H | 4-Cl-3-CF3 | 0.22 ± 0.02 | 0.11 ± 0.1 |
| 8f | H | 3-OCH3 | NA b | NA b |
| 8g | H | 4-OCH3 | 4.12 ± 0.27 | 27.24 ± 1.98 |
| 8h | OCH3 | H | NA b | NA b |
| 8i | OCH3 | 3-CF3 | 6.19 ± 0.54 | 5.80 ± 0.34 |
| 8j | OCH3 | 3-Cl | 10.9 ± 1.03 | 26.2 ± 2.17 |
| 8k | OCH3 | 4-Cl | 5.63 ± 0.36 | 3.45 ± 0.30 |
| 8l | OCH3 | 4-Cl-3-CF3 | 7.03 ± 0.61 | 21.43 ± 2.03 |
| 8m | OCH3 | 3-OCH3 | 23.02 ± 1.91 | 13.1 ± 1.12 |
| 8n | OCH3 | 4-OCH3 | 1.88 ± 0.12 | 0.80 ± 0.07 |
| Dox. | - | - | 1.93 ± 0.15 | 1.07 ± 0.07 |
| Sorafenib | - | - | 4.50 ± 0.30 | 1.71 ± 0.15 |
| Subpanel/Cell Line | Compound a | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 8b (NSC 793907) | 8d (NSC 793908) | 8e (NSC 793925) | 8i (NSC 793919) | 8j (NSC 793924) | 8l (NSC 793915) | 8m (NSC 793929) | 8n (NSC 793926) | ||
| Leukemia | CCRF-CEM | 58 | 19 | 71 | 50 | 32 | 45 | 11 | - |
| HL-60(TB) | 51 | 21 | 68 | 39 | 37 | 48 | 27 | 15 | |
| K-562 | 68 | 16 | 84 | 69 | 60 | 51 | 22 | 27 | |
| MOLT-4 | 67 | 28 | 76 | 61 | 58 | 57 | 35 | 15 | |
| RPMI-8226 | 78 | 10 | 91 | 66 | 49 | 57 | 26 | 17 | |
| SR | 64 | 57 | 65 | 49 | 48 | 40 | 18 | 43 | |
| Non-Small Cell Lung Cancer | A549/ATCC | 58 | 32 | 62 | 40 | 39 | 56 | 10 | 43 |
| EKVX | 59 | - | 51 | 34 | 17 | 33 | - | - | |
| HOP-62 | 33 | 19 | 38 | - | 23 | 23 | - | 44 | |
| HOP-92 | 32 | 19 | 73 | 45 | 40 | 30 | 19 | 27 | |
| NCI-H226 | 42 | - | 35 | 28 | - | 28 | - | 42 | |
| NCI-H23 | 53 | - | 70 | 29 | 11 | 29 | 10 | 33 | |
| NCI-H322M | 36 | 35 | 29 | - | 15 | 13 | - | 14 | |
| NCI-H460 | 52 | 36 | 70 | 39 | 17 | 48 | - | 83 | |
| NCI-H522 | 72 | 33 | 76 | 60 | 41 | 57 | 47 | 51 | |
| Colon Cancer | COLO 205 | 16 | - | 20 | 12 | - | - | - | - |
| HCC-2998 | 19 | - | 31 | 16 | 12 | - | - | 17 | |
| HCT-116 | 74 | 24 | 77 | 83 | 37 | 55 | 22 | 51 | |
| HCT-15 | 60 | 15 | 63 | 52 | 40 | 51 | - | - | |
| HT29 | 53 | 15 | 54 | 41 | 26 | 44 | 25 | 32 | |
| KM12 | 55 | - | 63 | 34 | 22 | 30 | - | 55 | |
| SW-620 | 19 | 23 | 33 | - | - | 10 | - | 36 | |
| CNS Cancer | SF-268 | 26 | - | 27 | 25 | 13 | 25 | - | 41 |
| SF-295 | 53 | - | 54 | 35 | 17 | 29 | - | - | |
| SF-539 | 20 | 16 | 19 | 23 | 11 | 13 | - | 30 | |
| SNB-19 | 33 | - | 43 | 22 | - | 22 | - | 29 | |
| SNB-75 | 23 | 16 | 27 | 34 | 16 | 20 | - | 53 | |
| U251 | 50 | 24 | 67 | 37 | 17 | 45 | - | 78 | |
| Melanoma | LOX IMVI | 53 | 15 | 36 | 30 | 20 | 22 | - | 12 |
| MALME-3M | 10 | - | 18 | - | - | 10 | - | 56 | |
| M14 | 63 | 14 | 69 | 69 | 50 | 53 | 10 | 28 | |
| MDA-MB-435 | 31 | 12 | 29 | 27 | 13 | 23 | - | 35 | |
| SK-MEL-2 | 25 | - | 38 | 27 | 16 | 13 | 13 | 21 | |
| SK-MEL-28 | 31 | 11 | 29 | 25 | 21 | 27 | - | 14 | |
| SK-MEL-5 | 59 | 13 | 45 | 42 | 32 | 37 | 16 | 78 | |
| UACC-257 | 39 | 22 | 43 | 19 | 29 | 44 | - | 45 | |
| UACC-62 | 37 | 29 | 35 | 19 | 25 | 23 | - | 41 | |
| Ovarian Cancer | IGROV1 | - | 17 | 48 | - | - | 17 | - | 23 |
| OVCAR-3 | 59 | - | 73 | 31 | 10 | 43 | - | - | |
| OVCAR-4 | 58 | 26 | 55 | 41 | - | 25 | - | 71 | |
| OVCAR-5 | 17 | - | 11 | - | 12 | 15 | - | - | |
| OVCAR-8 | 39 | - | 43 | 19 | 10 | 19 | - | 53 | |
| NCI/ADR-RES | 47 | - | 51 | 34 | 16 | 31 | - | 16 | |
| SK-OV-3 | 18 | 18 | 32 | - | 17 | 16 | - | 26 | |
| Renal Cancer | 786-0 | 23 | 10 | 44 | 31 | 29 | 25 | - | 89 |
| A498 | 48 | 21 | 43 | 39 | 31 | 43 | 15 | 29 | |
| RXF 393 | 15 | - | 19 | 23 | 18 | 28 | - | 42 | |
| SN12C | 33 | - | 43 | 21 | 14 | 22 | - | 36 | |
| TK-10 | 24 | - | 35 | 21 | - | 12 | - | 40 | |
| UO-31 | 45 | 11 | 61 | 26 | 21 | 28 | 20 | 19 | |
| Prostate | PC-3 | 76 | 24 | 86 | 73 | 61 | 67 | 41 | 19 |
| DU-145 | 32 | - | 46 | 18 | - | 20 | - | 28 | |
| Breast Cancer | MCF7 | 59 | 26 | 64 | 38 | 25 | 42 | 10 | 35 |
| MDA-MB-231 | 32 | - | 15 | 31 | 13 | 12 | - | 60 | |
| HS 578T | 12 | - | 24 | 26 | - | 19 | - | 61 | |
| BT-549 | 34 | - | 34 | 29 | - | 12 | 13 | 50 | |
| T-47D | 74 | 28 | 79 | 47 | 51 | 51 | 19 | 40 | |
| MDA-MB-468 | 73 | - | 62 | 51 | 29 | 52 | - | 17 | |
| Compound | IC50 (μM) a |
|---|---|
| WI-38 | |
| 8b | 91.5 ± 4.53 |
| 8e | 83.14 ± 5.21 |
| Compound | IC50 (μM) a |
|---|---|
| VEGFR-2 | |
| 8b | 5.00 ± 1.91 |
| 8e | 3.93 ± 0.73 |
| Sorafenib | 0.09 ± 0.01 |
| Compound | ADMET Solubility 1 | ADMET Solubility Level 2 | ADMET Absorption Level 3 | ADMET BBB 4 | ADMET BBB Level 4 |
|---|---|---|---|---|---|
| 8a | −4.499 | 2 | 0 | −0.045 | 2 |
| 8b | −5.93 | 2 | 0 | 0.247 | 1 |
| 8e | −6.698 | 1 | 0 | 0.452 | 1 |
| 8g | −4.554 | 2 | 0 | −0.191 | 2 |
| 8i | −5.971 | 2 | 0 | 0.10 | 1 |
| 8l | −6.723 | 1 | 0 | 0.306 | 1 |
| 8n | −4.623 | 2 | 0 | −0.337 | 2 |
| Compound | H-Bond Donor 1,* | H-Bond Acceptor 2,* | Molecular Weight 3 | ALogP 4 | No. of Rotatable Bond 5 | Polar Surface Area 6 (Å2) |
|---|---|---|---|---|---|---|
| 8a | 2 | 3 | 333.38 | 3.585 | 4 | 63.25 |
| 8b | 2 | 3 | 401.38 | 4.527 | 5 | 63.25 |
| 8e | 2 | 3 | 435.82 | 5.192 | 5 | 63.25 |
| 8g | 2 | 4 | 363.41 | 3.569 | 5 | 72.48 |
| 8i | 2 | 4 | 431.41 | 4.511 | 6 | 72.48 |
| 8l | 2 | 4 | 465.85 | 5.175 | 6 | 72.48 |
| 8n | 2 | 5 | 393.44 | 3.552 | 6 | 81.71 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Naggar, M.; Almahli, H.; Ibrahim, H.S.; Eldehna, W.M.; Abdel-Aziz, H.A. Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation. Molecules 2018, 23, 1459. https://doi.org/10.3390/molecules23061459
El-Naggar M, Almahli H, Ibrahim HS, Eldehna WM, Abdel-Aziz HA. Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation. Molecules. 2018; 23(6):1459. https://doi.org/10.3390/molecules23061459
Chicago/Turabian StyleEl-Naggar, Mohamed, Hadia Almahli, Hany S. Ibrahim, Wagdy M. Eldehna, and Hatem A. Abdel-Aziz. 2018. "Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation" Molecules 23, no. 6: 1459. https://doi.org/10.3390/molecules23061459
APA StyleEl-Naggar, M., Almahli, H., Ibrahim, H. S., Eldehna, W. M., & Abdel-Aziz, H. A. (2018). Pyridine-Ureas as Potential Anticancer Agents: Synthesis and In Vitro Biological Evaluation. Molecules, 23(6), 1459. https://doi.org/10.3390/molecules23061459