Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile



Abstract
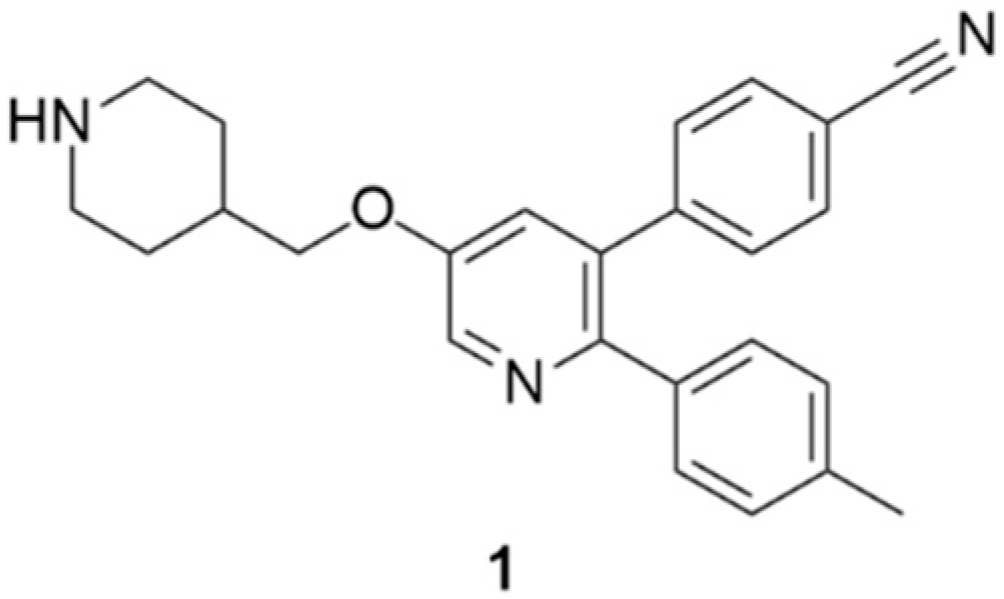
:
1. Introduction
2. Results
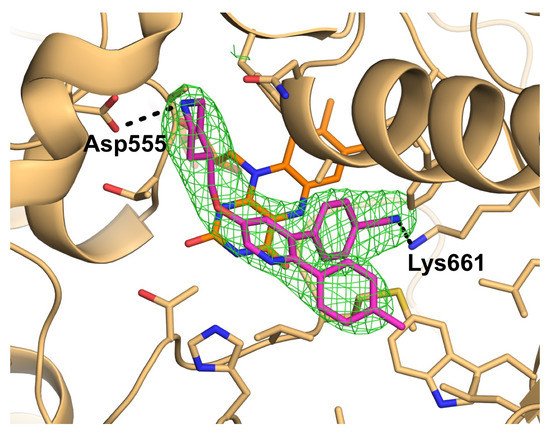
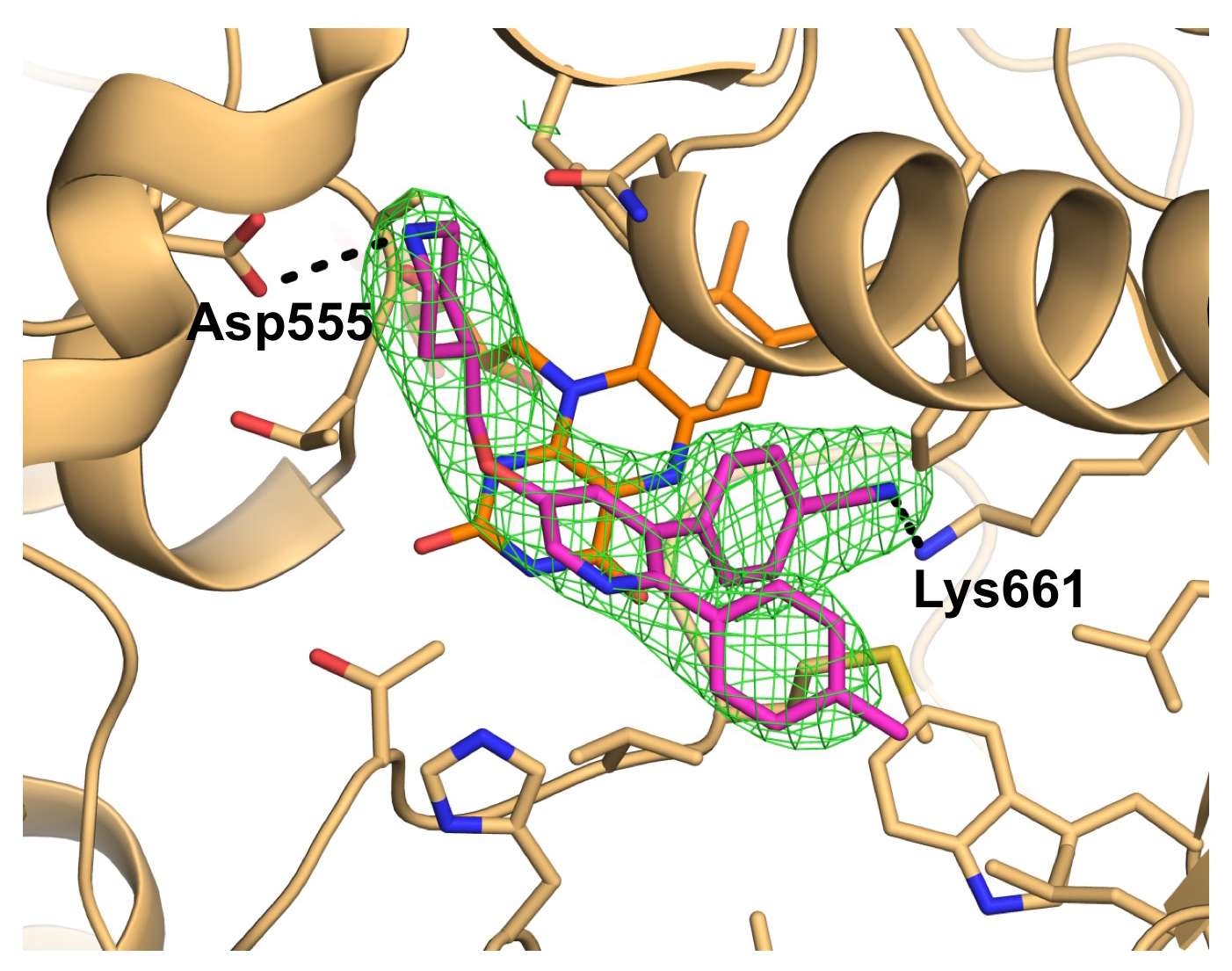
2.1. Overall Structure of LSD1•CoREST in Complex with 1
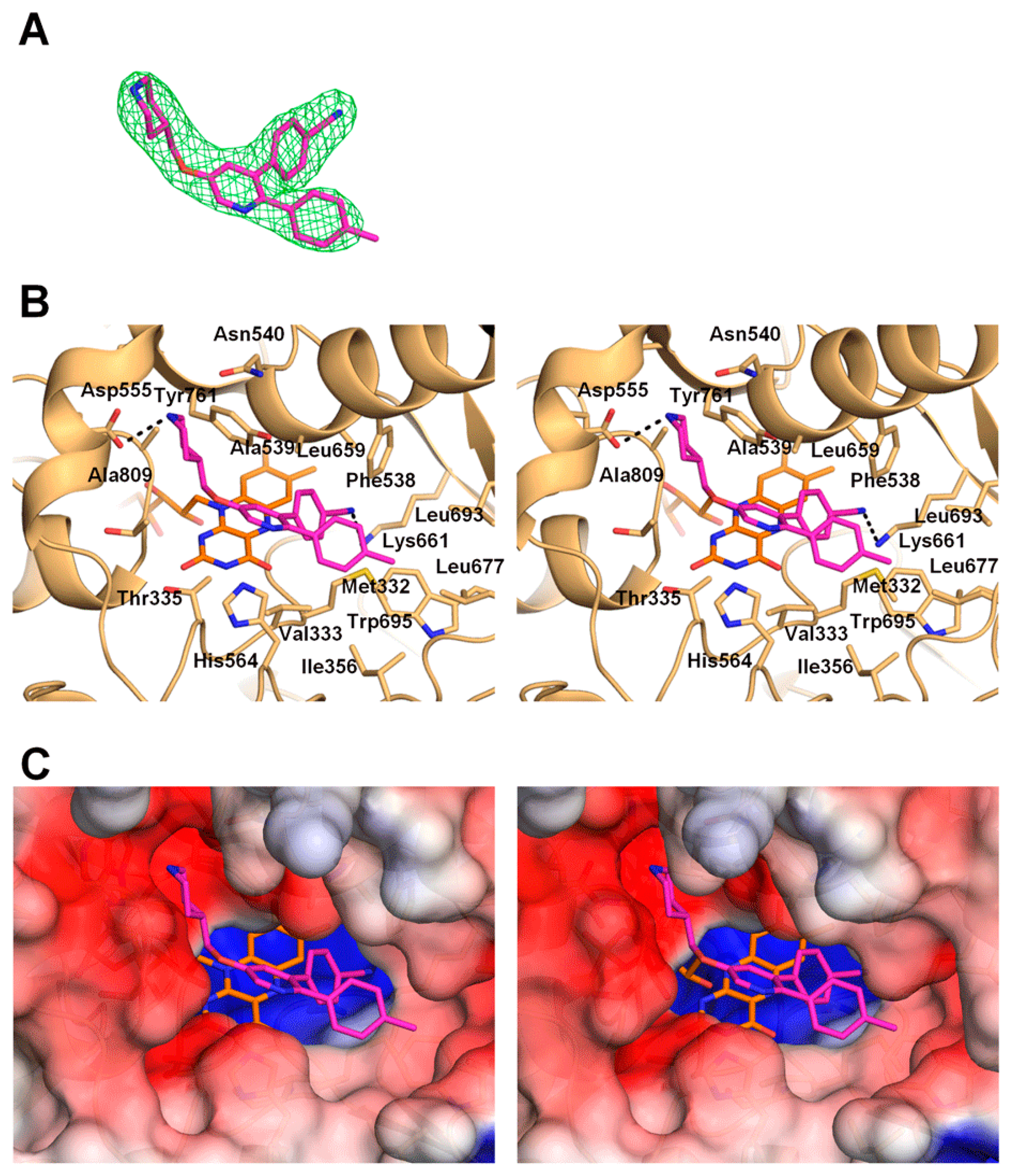
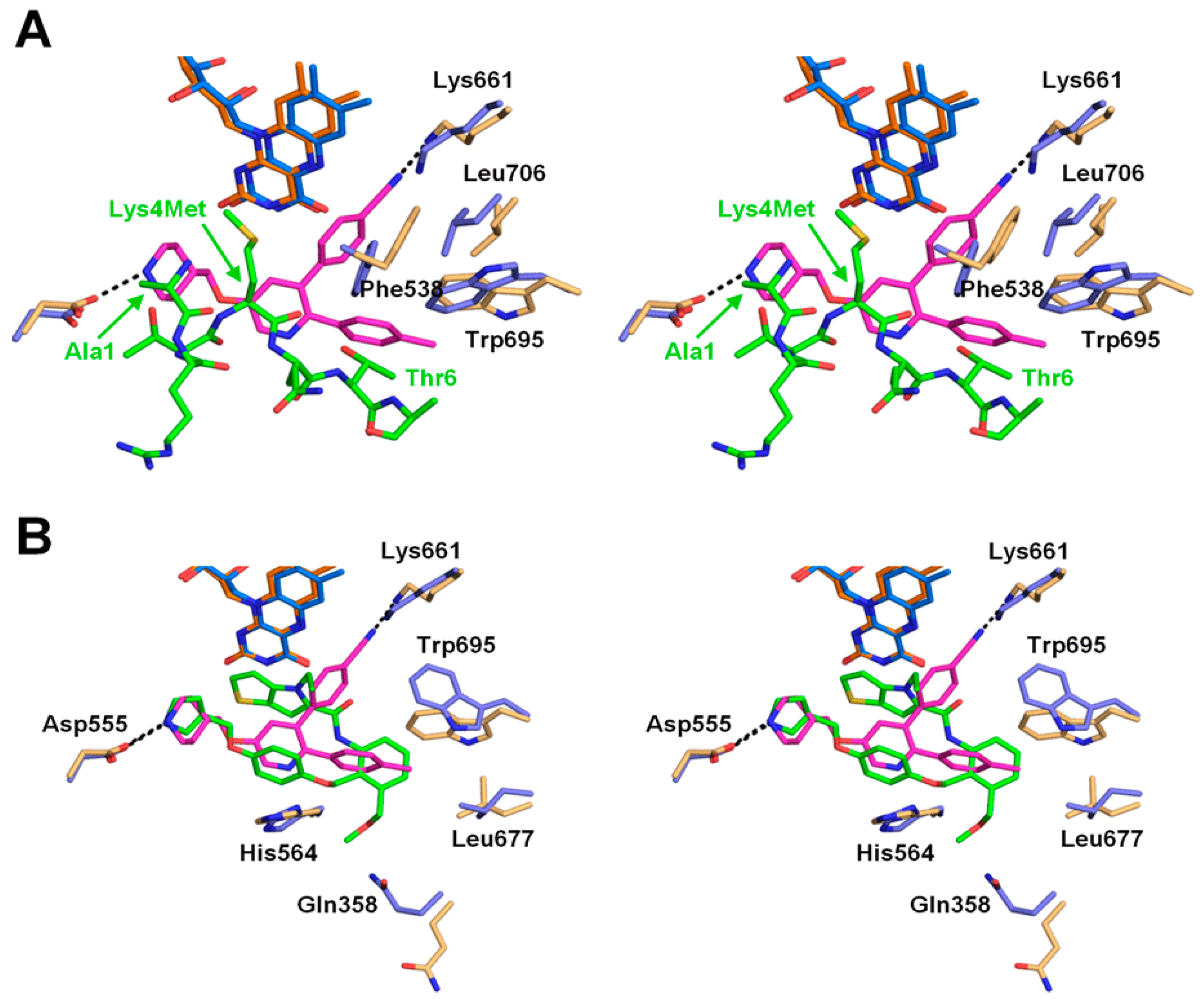
2.2. Interactions between LSD1 and 1
2.3. Comparison of the LSD1•CoREST•1 Structure with that of LSD1•CoREST-Bound H3 Peptide
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis of 1
4.3. Protein Preparation and Crystallization
4.4. X-ray Diffraction Data Collection and Structure Determination
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Koyama, D.; Kikuchi, J.; Honda, H.; Furukawa, Y. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015, 125, 3731–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnesecchi, J.; Forcet, C.; Zhang, L.; Tribollet, V.; Barenton, B.; Boudra, R.; Cerutti, C.; Billas, I.M.; Serandour, A.A.; Carroll, J.S.; et al. ERRα induces H3K9 demethylation by LSD1 to promote cell invasion. Proc. Natl. Acad. Sci. USA 2017, 114, 3909–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, H.; Umehara, T. Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 2017, 12, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Schenk, T.; Chen, W.C.; Gollner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Mimasu, S.; Umezawa, N.; Sato, S.; Higuchi, T.; Umehara, T.; Yokoyama, S. Structurally designed trans-2-phenylcyclopropylamine derivatives potently inhibit histone demethylase LSD1/KDM1. Biochemistry 2010, 49, 6494–6503. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; Karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. [Google Scholar] [CrossRef] [PubMed]
- Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. LSD1 inhibitors: A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.P.; McGonagle, A.E.; Wiseman, D.H.; Williams, E.L.; Jordan, A.M. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: Clinical significance and progress to date. Med. Res. Rev. 2015, 35, 586–618. [Google Scholar] [CrossRef] [PubMed]
- Speranzini, V.; Rotili, D.; Ciossani, G.; Pilotto, S.; Marrocco, B.; Forgione, M.; Lucidi, A.; Forneris, F.; Mehdipour, P.; Velankar, S.; et al. Polymyxins and quinazolines are LSD1/KDM1A inhibitors with unusual structural features. Sci. Adv. 2016, 2, e1601017. [Google Scholar] [CrossRef] [PubMed]
- Sartori, L.; Mercurio, C.; Amigoni, F.; Cappa, A.; Faga, G.; Fattori, R.; Legnaghi, E.; Ciossani, G.; Mattevi, A.; Meroni, G.; et al. Thieno[3,2-b]pyrrole-5-carboxamides as new reversible inhibitors of histone lysine demethylase KDM1A/LSD1. Part 1: High-throughput screening and preliminary exploration. J. Med. Chem. 2017, 60, 1673–1692. [Google Scholar] [CrossRef] [PubMed]
- Vianello, P.; Sartori, L.; Amigoni, F.; Cappa, A.; Faga, G.; Fattori, R.; Legnaghi, E.; Ciossani, G.; Mattevi, A.; Meroni, G.; et al. Thieno[3,2-b]pyrrole-5-carboxamides as new reversible inhibitors of histone lysine demethylase KDM1A/LSD1. Part 2: Structure-based drug design and structure-activity relationship. J. Med. Chem. 2017, 60, 1693–1715. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z.; Pakhomova, S.; Loukachevitch, L.V.; Calcutt, M.W.; Newcomer, M.E.; Wagner, C. Crystal structure of the histone lysine specific demethylase LSD1 complexed with tetrahydrofolate. Protein Sci. 2014, 23, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhou, C.; Yao, Y.; Wei, L.; Feng, Z.; Deng, L.; Song, Y. 3-(Piperidin-4-ylmethoxy)pyridine containing compounds are potent inhibitors of lysine specific demethylase 1. J. Med. Chem. 2016, 59, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Forneris, F.; Binda, C.; Adamo, A.; Battaglioli, E.; Mattevi, A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J. Biol. Chem. 2007, 282, 20070–20074. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. USA 2006, 103, 13956–13961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mould, D.P.; Alli, C.; Bremberg, U.; Cartic, S.; Jordan, A.M.; Geitmann, M.; Maiques-Diaz, A.; McGonagle, A.E.; Somervaille, T.C.P.; Spencer, G.J.; et al. Development of (4-cyanophenyl)glycine derivatives as reversible inhibitors of lysine specific demethylase 1. J. Med. Chem. 2017, 60, 7984–7999. [Google Scholar] [CrossRef] [PubMed]
- Amano, Y.; Kikuchi, M.; Sato, S.; Yokoyama, S.; Umehara, T.; Umezawa, N.; Higuchi, T. Development and crystallographic evaluation of histone H3 peptide with N-terminal serine substitution as a potent inhibitor of lysine-specific demethylase 1. Bioorg. Med. Chem. 2017, 25, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, O.S.; Womack, T.O.; Sharff, A.; Flensburg, C.; Keller, P.; Paciorek, W.; Vonrhein, C.; Bricogne, G. Grade, version 1.2.13; Global Phasing Ltd.: Cambridge, UK, 2011. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, version 1.8.4.0; Schrödinger, LLC: New York, NY, USA, 2016.
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | 5YJB |
|---|---|
| Data Collection | |
| Beamline | SPring-8 BL26B2 |
| Wavelength (Å) | 1.000 |
| Space group | I222 |
| Unit Cell Dimensions | |
| a, b, c (Å) | 121.1, 179.6, 235.0 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 48.82−2.96 (3.05−2.96) * |
| No. of unique reflections | 53,652 |
| Completeness (%) | 100 (100) |
| Multiplicity | 7.5 (7.6) |
| Mean I/σ (I) | 16.2 (1.7) |
| Rsym (%) | 11.4 (142) |
| Rpim (%) | 4.4 (54.8) |
| CC1/2 | 0.999 (0.747) |
| Refinement | |
| Rwork (%) | 18.9 |
| Rfree (%) | 21.9 |
| RMSD bond lengths (Å) | 0.008 |
| RMSD bond angles (°) | 0.945 |
| No. of reflections (total) | 53,614 |
| No. of reflections (test set) | 1053 |
| No. of Atoms | |
| Total | 6313 |
| Protein | 6219 |
| FAD | 53 |
| Inhibitor | 29 |
| Glycerol | 12 |
| Mean B-factor (Å2) | |
| Overall | 92.9 |
| Protein | 93.2 |
| FAD | 67.8 |
| Inhibitor | 66.6 |
| Glycerol | 82.9 |
| Ramachandran Plot (%) | |
| Favored | 96.5 |
| Allowed | 3.5 |
| Molprobity clashscore | 4.6 (100th percentile) |
| Molprobity score | 1.45 (100th percentile) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niwa, H.; Sato, S.; Hashimoto, T.; Matsuno, K.; Umehara, T. Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile. Molecules 2018, 23, 1538. https://doi.org/10.3390/molecules23071538
Niwa H, Sato S, Hashimoto T, Matsuno K, Umehara T. Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile. Molecules. 2018; 23(7):1538. https://doi.org/10.3390/molecules23071538
Chicago/Turabian StyleNiwa, Hideaki, Shin Sato, Tomoko Hashimoto, Kenji Matsuno, and Takashi Umehara. 2018. "Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile" Molecules 23, no. 7: 1538. https://doi.org/10.3390/molecules23071538
APA StyleNiwa, H., Sato, S., Hashimoto, T., Matsuno, K., & Umehara, T. (2018). Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl)pyridin-3-yl]benzonitrile. Molecules, 23(7), 1538. https://doi.org/10.3390/molecules23071538