Theoretical Model of EphA2-Ephrin A1 Inhibition


Abstract
:1. Introduction
2. Results and Discussion
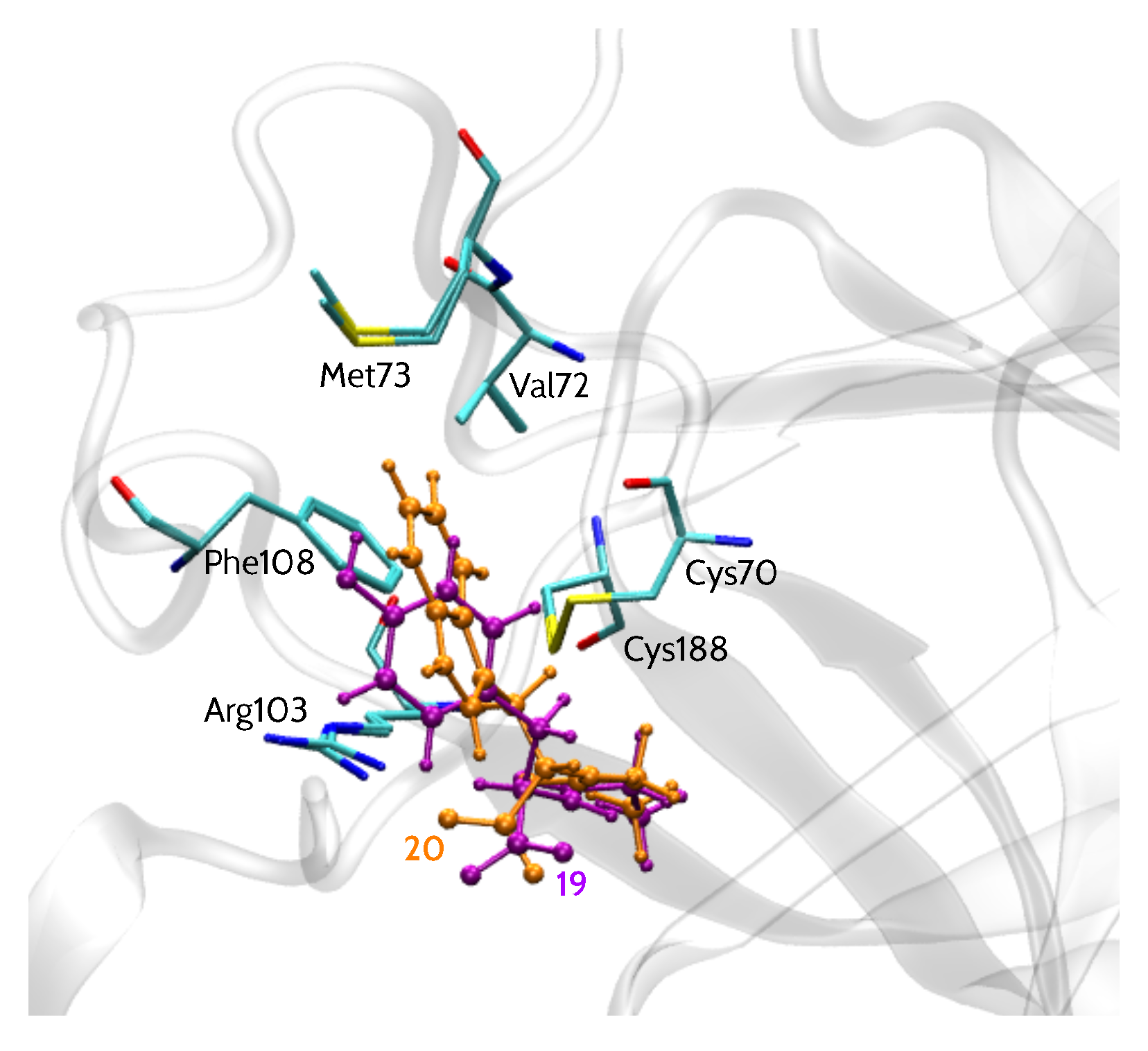
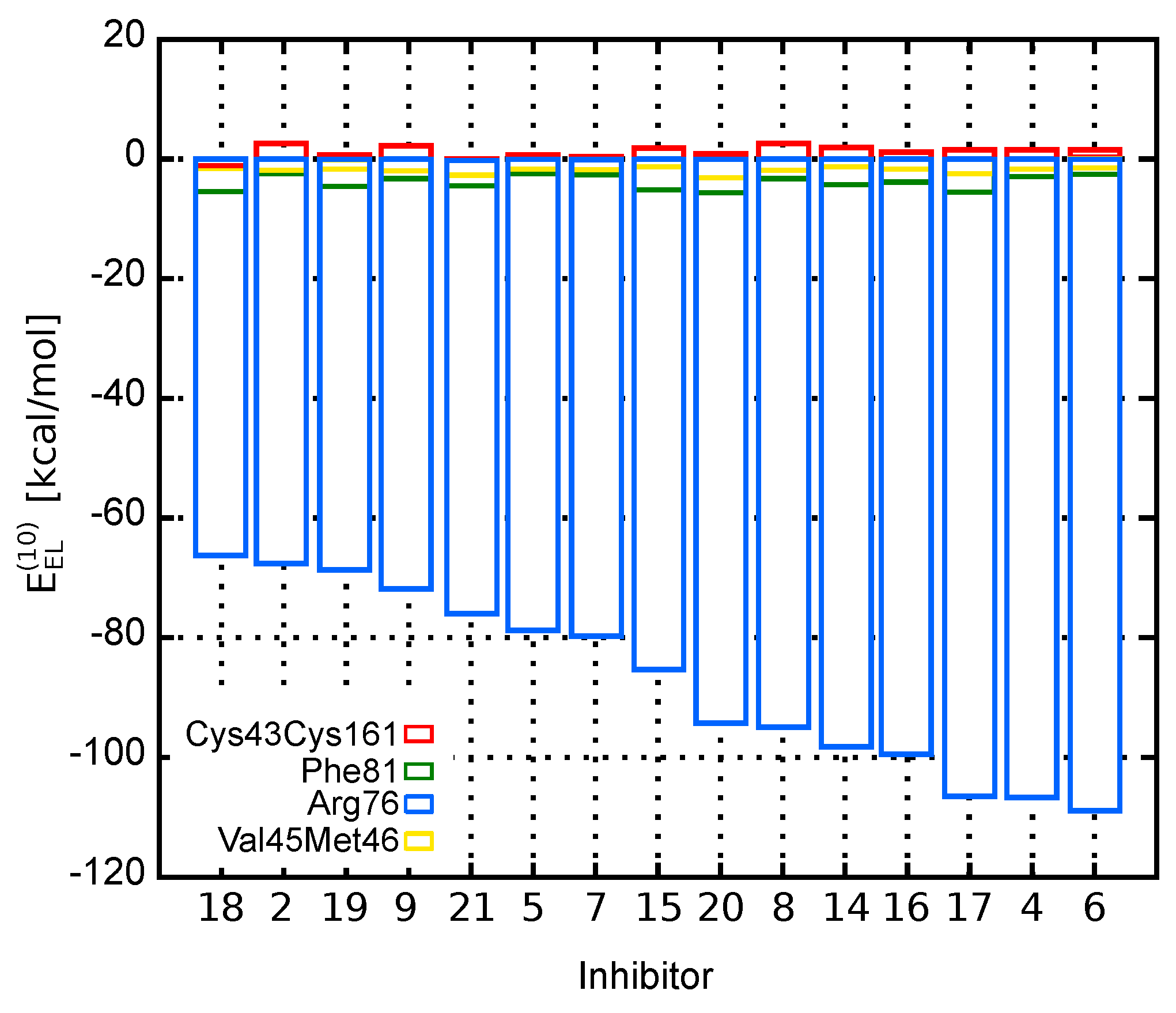
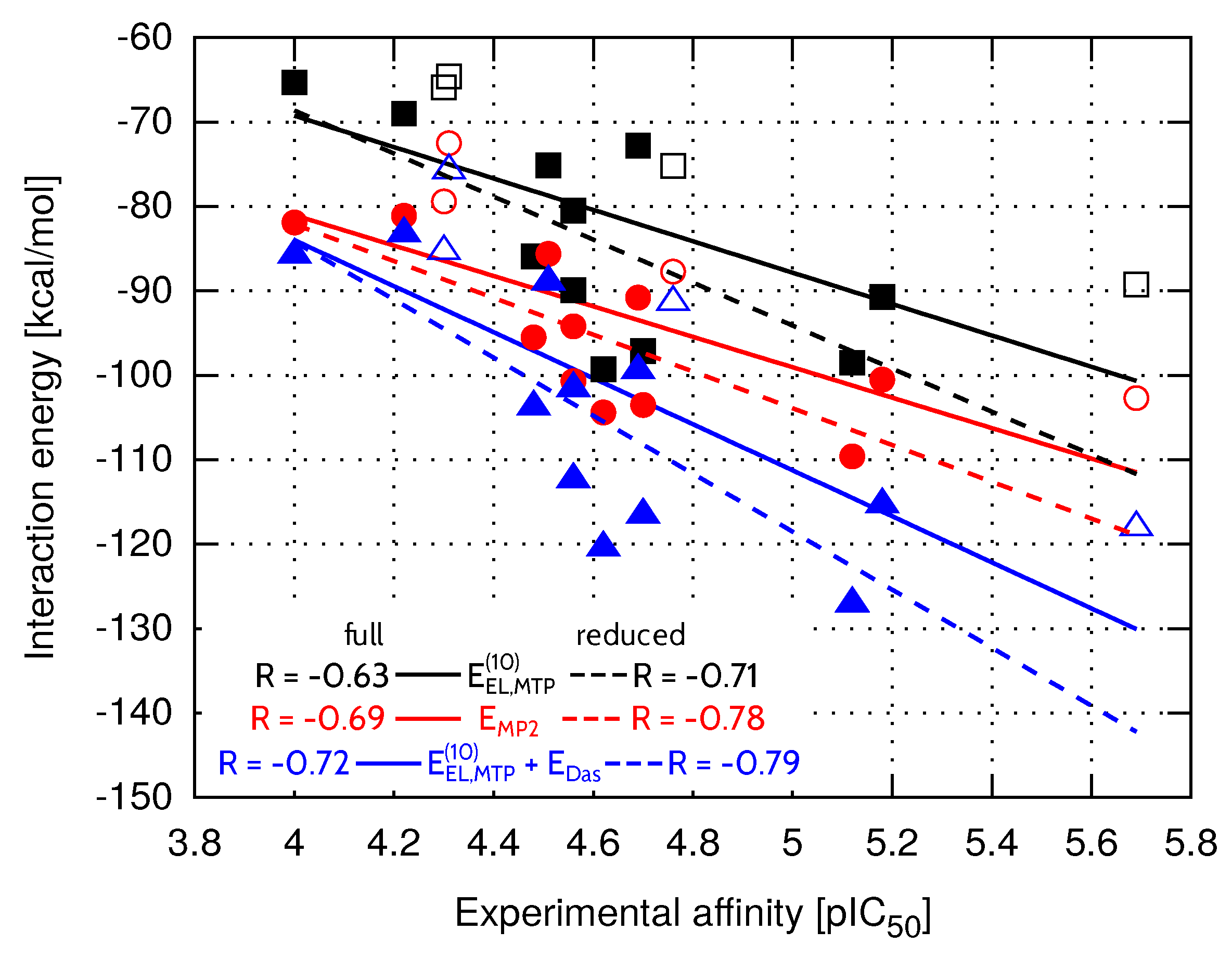
2.1. Theoretical Models
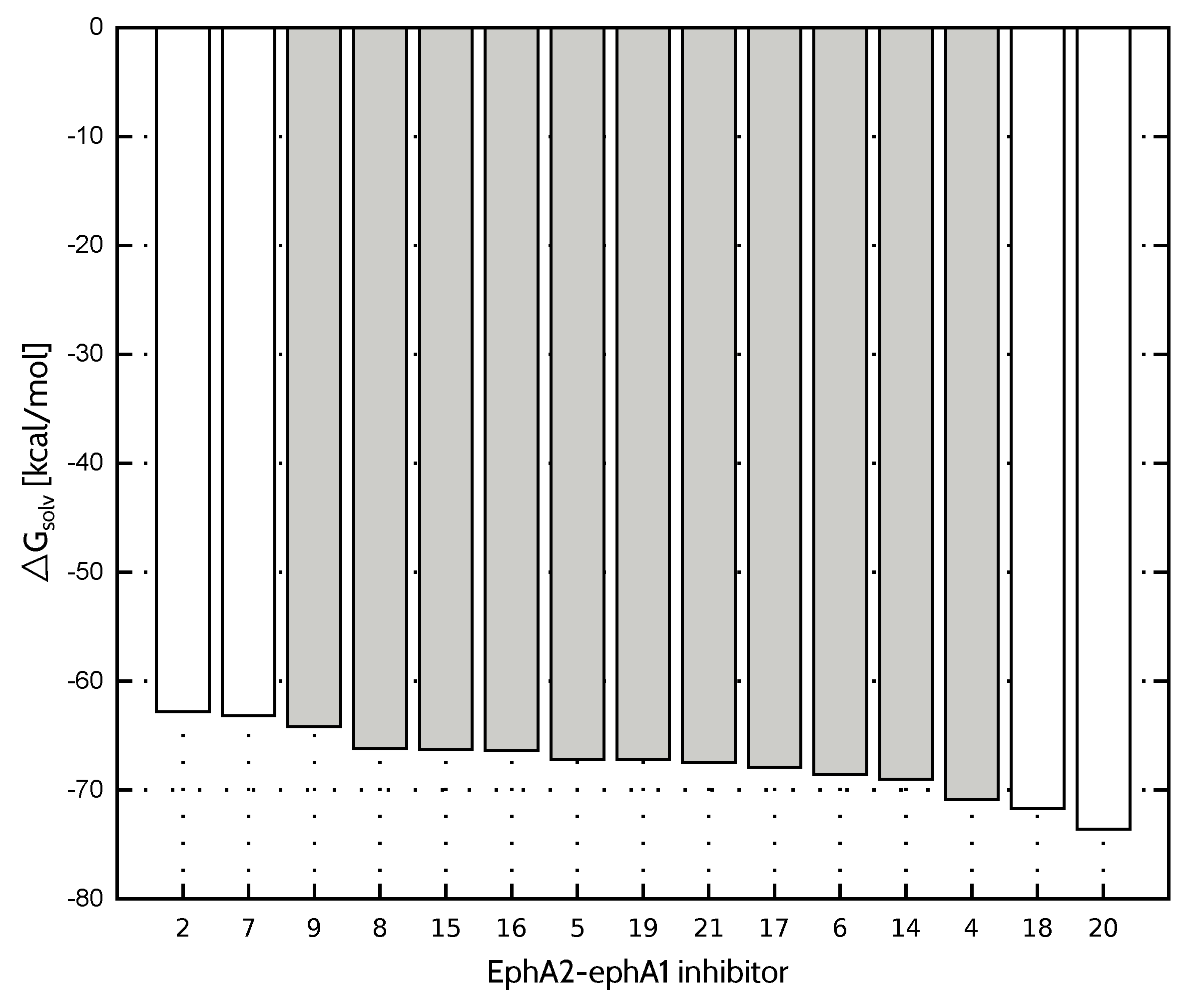
2.2. Solvation Energy of Inhibitors
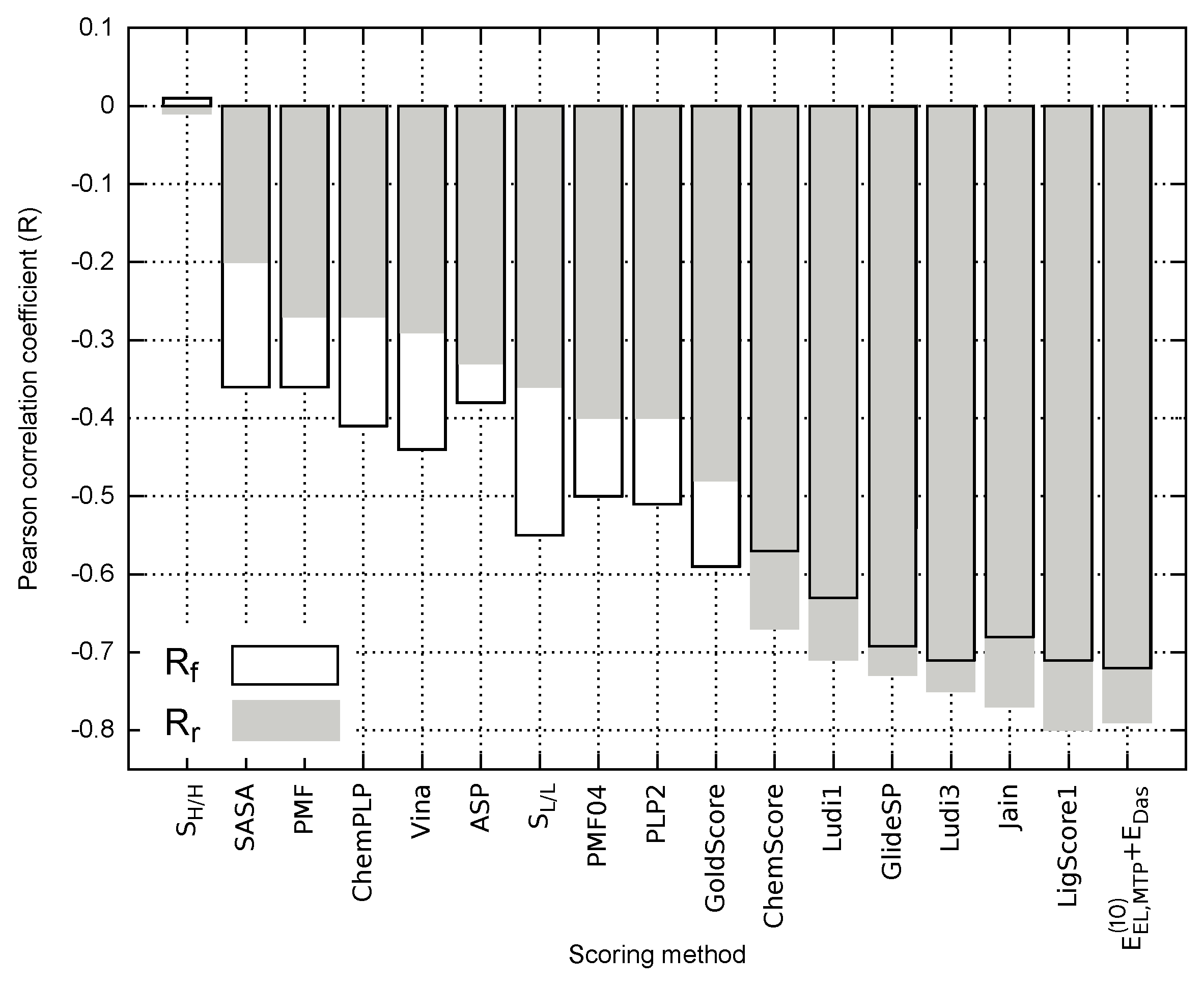
2.3. Empirical Evaluation of EphA2-Ephrin A1 Inhibitors
3. Materials and Methods
3.1. Preparation of the Structures
3.2. Interaction Energy Calculations
3.3. Solvation Energy Calculations
3.4. Empirical Scoring
3.5. Evaluation of the Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lisle, J.E.; Mertens-Walker, I.; Rutkowski, R.; Herington, A.C.; Stephenson, S.A. Eph receptors and their ligands: Promising molecular biomarkers and therapeutic targets in prostate cancer. BBA-Rev. Cancer 2013, 1835, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Tognolini, M.; Hassan-Mohamed, I.; Giorgio, C.; Zanotti, I.; Lodola, A. Therapeutic perspectives of Eph-ephrin system modulation. Drug Discov. Today 2014, 19, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Chumley, M.J.; Lackmann, M.; Li, C.; Barton, W.A.; Jeffrey, P.D.; Vearing, C.; Geleick, D.; Feldheim, D.A.; Boyd, A.W.; et al. Repelling class discrimination: Ephrin-A5 binds to and activates EphB2 receptor signaling. Nat. Neurosci. 2004, 7, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Noberini, R.; Huan, X.; Shi, J.; Pasquale, E.B.; Song, J. Structural Characterization of the EphA4-Ephrin-B2 Complex Reveals New Features Enabling Eph-Ephrin Binding Promiscuity. J. Biol. Chem. 2010, 285, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Son, A.I.; Zhou, R. Roles of EphA2 in Development and Disease. Genes 2013, 4, 334–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavent, M.; Seiradake, E.; Jones, E.Y.; Sansom, M.S.P. Structures of the EphA2 Receptor at the Membrane: Role of Lipid Interactions. Structure 2016, 24, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, C.; Mohamed, I.H.; Flammini, L.; Barocelli, E.; Incerti, M.; Lodola, A.; Tognolini, M. Lithocholic Acid Is an Eph-ephrin Ligand Interfering with Eph-kinase Activation. PLoS ONE 2011, 6, e18128. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Kato, H.; Fukuchi, M.; Nakajima, M.; Kuwano, H. EphA2 overexpression correlates with poor prognosis in esophageal squamous cell carcinoma. Int. J. Cancer 2003, 103, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, D.B.; Xu, K.; Himanen, J.P. Eph/ephrin recognition and the role of Eph/ephrin clusters in signaling initiation. BBA-Proteins Proteom. 2013, 1834, 2160–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, S.; Incerti, M.; Tognolini, M.; Castelli, R.; Pala, D.; Hassan-Mohamed, I.; Giorgio, C.; De Franco, F.; Gioiello, A.; Vicini, P.; et al. Synthesis and Structure-Activity Relationships of Amino Acid Conjugates of Cholanic Acid as Antagonists of the EphA2 Receptor. Molecules 2013, 18, 13043–13060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noberini, R.; Koolpe, M.; Peddibhotla, S.; Dahl, R.; Su, Y.; Cosford, N.D.P.; Roth, G.P.; Pasquale, E.B. Small Molecules Can Selectively Inhibit Ephrin Binding to the EphA4 and EphA2 Receptors. J. Biol. Chem. 2008, 283, 29461–29472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, A.; Myshkin, E.; Qin, H.; Guo, H.; Miao, H.; Tochtrop, G.P.; Hsieh, J.T.; Page, P.; Liu, L.; Lindner, D.J.; et al. A Small Molecule Agonist of EphA2 Receptor Tyrosine Kinase Inhibits Tumor Cell Migration In Vitro and Prostate Cancer Metastasis In Vivo. PLoS ONE 2012, 7, e42120. [Google Scholar] [CrossRef] [PubMed]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino Acid Conjugates of Lithocholic Acid as Antagonists of the EphA2 Receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Callegari, D.; Incerti, M.; Pala, D.; Giorgio, C.; Brunetti, J.; Bracci, L.; Vicini, P.; Barocelli, E.; Capoferri, L.; et al. Exploiting Free-Energy Minima to Design Novel EphA2 Protein-Protein Antagonists: From Simulation to Experiment and Return. Chem. Eur. J. 2016, 22, 8048–8052. [Google Scholar] [CrossRef] [PubMed]
- Tognolini, M.; Incerti, M.; Hassan-Mohamed, I.; Giorgio, C.; Russo, S.; Bruni, R.; Lelli, B.; Bracci, L.; Noberini, R.; Pasquale, E.B.; et al. Structure-Activity Relationships and Mechanism of Action of Eph-ephrin Antagonists: Interaction of Cholanic Acid with the EphA2 Receptor. ChemMedChem 2012, 7, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.R.; Pisabarro, M.T.; Gago, F.; Wade, R.C. Prediction of drug binding affinities by computer binding analysis. J. Med. Chem. 1995, 38, 2681–2691. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of Protein-Ligand Interactions. Docking and Scoring: Successes and Gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar] [CrossRef] [PubMed]
- Doweyko, A.M. 3D-QSAR Illusions. J. Comput.-Aided Mol. Des. 2004, 18, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Yilmazer, N.D.; Korth, M. Comparison of Molecular Mechanics, Semi-Empirical Quantum Mechanical, and Density Functional Theory Methods for Scoring Protein-Ligand Interactions. J. Phys. Chem. B 2013, 117, 8075–8084. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienstock, R.J. Computational drug design targeting protein–protein interactions. Curr. Pharm. Des. 2012, 18, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Laraia, L.; McKenzie, G.; Spring, D.R.; Venkitaraman, A.R.; Huggins, D.J. Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting Protein-Protein Interactions. Chem. Biol. 2015, 22, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Kuenemann, M.A.; Sperandio, O.; Labbé, C.M.; Lagorce, D.; Miteva, M.A.; Villoutreix, B.O. In silico design of low molecular weight protein–protein interaction inhibitors: Overall concept and recent advances. Prog. Biophys. Mol. Biol. 2015, 119, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of Potent Keap1-Nrf2 Protein-Protein Interaction Inhibitor Based on Molecular Binding Determinants Analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug design targeting protein–protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: Discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Zhang, Q.; Chen, K.; Zhu, W. Probing Origin of Binding Difference of inhibitors to MDM2 and MDMX by Polarizable Molecular Dynamics Simulation and QM/MM-GBSA Calculation. Sci. Rep. 2015, 5, 17421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Cai, L.; Chen, C.; Xie, X.; Zhao, Q.; Zhao, X.; Zhou, H.Y.; Han, B.; Peng, C. Computational analysis of spiro-oxindole inhibitors of the MDM2-p53 interaction: Insights and selection of novel inhibitors. J. Biomol. Struct. Dyn. 2016, 34, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Dyguda, E.; Grembecka, J.; Sokalski, W.A.; Leszczyński, J. Origins of the activity of PAL and LAP enzyme inhibitors: Towards ab initio binding affinity prediction. J. Am. Chem. Soc. 2005, 127, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, R.; Dyguda-Kazimierowicz, E.; Sieńczyk, M.; Feliks, M.; Sokalski, W.A.; Oleksyszyn, J. The molecular basis of urokinase inhibition: From the nonempirical analysis of intermolecular interactions to the prediction of binding affinity. J. Mol. Model. 2007, 13, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef] [PubMed]
- Podeszwa, R.; Pernal, K.; Patkowski, K.; Szalewicz, K. Extension of the Hartree-Fock Plus Dispersion Method by First-Order Correlation Effects. J. Phys. Chem. Lett. 2010, 1, 550–555. [Google Scholar] [CrossRef]
- Pernal, K.; Podeszwa, R.; Patkowski, K.; Szalewicz, K. Dispersionless Density Functional Theory. Phys. Rev. Lett. 2009, 103, 263201. [Google Scholar] [CrossRef] [PubMed]
- Giedroyć-Piasecka, W.; Dyguda-Kazimierowicz, E.; Beker, W.; Mor, M.; Lodola, A.; Sokalski, W.A. Physical Nature of Fatty Acid Amide Hydrolase Interactions with Its Inhibitors: Testing a Simple Nonempirical Scoring Model. J. Phys. Chem. B 2014, 118, 14727–14736. [Google Scholar] [CrossRef] [PubMed]
- Jedwabny, W.; Panecka-Hofman, J.; Dyguda-Kazimierowicz, E.; Wade, R.C.; Sokalski, W.A. Application of a simple quantum chemical approach to ligand fragment scoring for Trypanosoma brucei pteridine reductase 1 inhibition. J. Comput.-Aided Mol. Des. 2017, 31, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedwabny, W.; Kłossowski, S.; Purohit, T.; Cierpicki, T.; Grembecka, J.; Dyguda-Kazimierowicz, E. Theoretical models of inhibitory activity for inhibitors of protein–protein interactions: Targeting menin-mixed lineage leukemia with small molecules. Med. Chem. Commun. 2017, 8, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Ryde, U.; Söderhjelm, P. Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chem. Rev. 2016, 116, 5520–5566. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, T.; Okimoto, N.; Taiji, M. Assessment and Acceleration of Binding Energy Calculations for Protein-Ligand Complexes by the Fragment Molecular Orbital Method. J. Comput. Chem. 2015, 36, 2209–2218. [Google Scholar] [CrossRef] [PubMed]
- Tognolini, M.; Lodola, A. Targeting the Eph-ephrin System with Protein-Protein Interaction (PPI) Inhibitors. Curr. Drug Targets 2015, 16, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Sokalski, W.A.; Roszak, S.; Pecul, K. An efficient procedure for decomposition of the SCF interaction energy into components with reduced basis set dependence. Chem. Phys. Lett. 1988, 153, 153–159. [Google Scholar] [CrossRef]
- Góra, R.W.; Sokalski, W.A.; Leszczyński, J.; Pett, V. The nature of interactions in the ionic crystal of 3-pentenenitrile, 2-nitro-5-oxo, ion(-1) sodium. J. Phys. Chem. B 2005, 109, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Beker, W.; Langner, K.M.; Dyguda-Kazimierowicz, E.; Feliks, M.; Sokalski, W.A. Low-Cost Prediction of Relative Stabilities of Hydrogen-Bonded Complexes from Atomic Multipole Moments for Overly Short Intermolecular Distances. J. Comput. Chem. 2013, 34, 1797–1799. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Krammer, A.; Kirchhoff, P.D.; Jiang, X.; Venkatachalam, C.M.; Waldman, M. LigScore: A novel scoring function for predicting binding affinities. J. Mol. Graph. Model. 2005, 23, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.B.; Fogel, L.J.; Freer, S.T. Molecular Recognition of the Inhibitor AG-1343 by HIV-1 Protease: Conformationally Flexible Docking by Evolutionary Programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Bouzida, D.; Rejto, P.A. Rational Drug Design: Novel Methodology and Practical Applications; American Chemical Society: Washington, DC, USA, 1999. [Google Scholar]
- Jain, A.N. Scoring noncovalent protein–ligand interactions: A continuous differentiable function tuned to compute binding affinities. J. Comput. Aided Mol. Des. 1996, 10, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Martin, Y.C. A General and Fast Scoring Function for Protein-Ligand Interactions: A Simplified Potential Approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. PMF Scoring Revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from the de novo design or 3D database search programs. J. Comput. Aided Mol. Des. 1998, 12, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment; Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Li, J.; Han, L.; Liu, J.; Zhao, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model. 2014, 54, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.W.; Jorgensen, W.L. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 2000, 112, 8910–8922. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Stote, R.H.; Karplus, M. Zinc binding in proteins and solution: A simple but accurate nonbonded representation. Proteins 1995, 23, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestro Version 9.3; Schrödinger, LLC: New York, NY, USA, 2012.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 2002, 100, 65–73. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Selfconsistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cances, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct.-THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Pascualahuir, J.L.; Silla, E.; Tunon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Scalmani, G.; Frisch, M.J.; Barone, V. Toward effective and reliable fluorescence energies in solution by a new state specific polarizable continuum model time dependent density functional theory approach. J. Chem. Phys. 2007, 127, 074504. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Barone, V.; Scalmani, G.; Frisch, M.J. A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution. J. Chem. Phys. 2006, 125, 054103. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Englebienne, P.; Moitessier, N. Docking Ligands into Flexible and Solvated Macromolecules. 4. Are Popular Scoring Functions Accurate for this Class of Proteins? J. Chem. Inf. Model. 2009, 49, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation. Master’s Thesis, Computer Science Department, University of Missouri-Rolla, Rolla, MO, USA, 1998. [Google Scholar]
- Falsafi, S.; Karimi, Z. SASA.tcl. Available online: http://www.ks.uiuc.edu/Research/vmd/mailing_list/vmd-l/att-18670/sasa.tcl (accessed on 30 May 2018).
- PyMOL(TM) Molecular Graphics System, Version 1.7.0.0.; Schrödinger, LLC: New York, NY, USA, 2013.
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput.-Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger LLC. Schrödinger Release 2018-1, Glide; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Langner, K.M.; Beker, W.; Sokalski, W.A. Robust Predictive Power of the Electrostatic Term at Shortened Intermolecular Distances. J. Phys. Chem. Lett. 2012, 3, 2785–2789. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
| Inhibitor | X Substituent | pIC |
| 2 (Gly) |  | 4.31 |
| 4 (l-Ala) |  | 4.70 |
| 5 (d-Ala) |  | 4.51 |
| 6 (l-Val) |  | 4.62 |
| 7 (d-Val) |  | 4.76 |
| 8 (l-Ser) |  | 4.48 |
| 9 (d-Ser) |  | 4.22 |
| 14 (l-Met) |  | 4.56 |
| 15 (d-Met) |  | 4.56 |
| 16 (l-Phe) |  | 5.18 |
| 17 (d-Phe) |  | 5.12 |
| 18 (l-Tyr) |  | 4.30 |
| 19 (d-Tyr) |  | 4.00 |
| 20 (l-Trp) |  | 5.69 |
| 21 (d-Trp) |  | 4.69 |
| Inhibitor | pICb | ||||||
|---|---|---|---|---|---|---|---|
| 20 (l-Trp) | 5.69 | −89.2 | −101.3 | −66.5 | −83.5 | −102.7 | −118.0 |
| 16 (l-Phe) | 5.18 | −90.7 | −102.5 | −65.6 | −86.1 | −100.5 | −115.3 |
| 17 (d-Phe) | 5.12 | −98.5 | −111.4 | −70.1 | −92.6 | −109.6 | −127.0 |
| 7 (d-Val) | 4.76 | −75.2 | −83.3 | −65.7 | −77.4 | −87.7 | −91.3 |
| 4 (l-Ala) | 4.70 | −97.1 | −108.5 | −73.7 | −94.1 | −103.5 | −116.5 |
| 21 (d-Trp) | 4.69 | −72.8 | −82.3 | −57.9 | −70.9 | −90.8 | −99.4 |
| 6 (l-Val) | 4.62 | −99.3 | −110.0 | −71.9 | −94.4 | −104.4 | −120.4 |
| 14 (l-Met) | 4.56 | −89.9 | −101.1 | −69.1 | −87.7 | −100.7 | −112.3 |
| 15 (d-Met) | 4.56 | −80.5 | −89.5 | −67.3 | −80.6 | −94.2 | −101.5 |
| 5 (d-Ala) | 4.51 | −75.1 | −82.2 | −66.7 | −76.9 | −85.6 | −88.9 |
| 8 (l-Ser) | 4.48 | −85.9 | −96.6 | −70.4 | −86.2 | −95.5 | −103.7 |
| 2 (Gly) | 4.31 | −64.6 | −69.3 | −56.2 | −65.0 | −72.5 | −75.7 |
| 18 (l-Tyr) | 4.30 | −65.9 | −73.2 | −55.3 | −65.3 | −79.4 | −85.3 |
| 9 (d-Ser) | 4.22 | −69.0 | −74.7 | −62.6 | −71.4 | −81.1 | −83.2 |
| 19 (d-Tyr) | 4.00 | −65.3 | −74.1 | −55.8 | −66.5 | −81.9 | −85.7 |
| R c | −0.63 | −0.65 | −0.44 | −0.55 | −0.69 | −0.72 | |
| d | 75.0 | 76.9 | 65.4 | 69.2 | 75.0 | 77.9 | |
| SEe | 10.1 | 11.5 | 5.6 | 9.0 | 8.2 | 11.5 | |
| Inhibitor | pICb | |||
|---|---|---|---|---|
| 20 (l-Trp) | 5.69 | −73.6 | −81.2 | 7.6 |
| 16 (l-Phe) | 5.18 | −66.4 | −73.5 | 7.2 |
| 17 (d-Phe) | 5.12 | −67.9 | −75.3 | 7.4 |
| 7 (d-Val) | 4.76 | −63.2 | −70.0 | 6.8 |
| 4 (l-Ala) | 4.70 | −70.9 | −77.0 | 6.0 |
| 21 (d-Trp) | 4.69 | −67.5 | −75.2 | 7.7 |
| 6 (l-Val) | 4.62 | −68.6 | −75.5 | 7.0 |
| 14 (l-Met) | 4.56 | −69.0 | −75.9 | 6.9 |
| 15 (d-Met) | 4.56 | −66.3 | −73.5 | 7.2 |
| 5 (d-Ala) | 4.51 | −67.2 | −73.4 | 6.2 |
| 8 (l-Ser) | 4.48 | −66.2 | −72.0 | 5.8 |
| 2 (Gly) | 4.31 | −62.8 | −68.1 | 5.3 |
| 18 (l-Tyr) | 4.30 | −71.7 | −78.9 | 7.2 |
| 9 (d-Ser) | 4.22 | −64.2 | −70.2 | 6.0 |
| 19 (d-Tyr) | 4.00 | −67.2 | −74.9 | 7.7 |
| R c | −0.43 | −0.46 | 0.37 | |
| EphA2-Ephrin A1 | Menin-MLL | FAAH | TbPTR1 | |
|---|---|---|---|---|
| a | ||||
| b | ||||
| c |
| Scoring Function | FAAH a | menin-MLL b | EphA2-ephrin A1 c | |||
|---|---|---|---|---|---|---|
| R d | e | R | R | |||
| LigScore1 | 44.6 | 75.2 | 79.6 | |||
| Jain | 71.4 | 77.8 | 83.3 | |||
| PLP2 | 65.8 | 80.4 | 72.2 | |||
| Ludi1 | 73.2 | 58.8 | 75.9 | |||
| PMF | 77.1 | 41.2 | 66.7 | |||
| 74.9 | 81.1 | 79.6 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jedwabny, W.; Lodola, A.; Dyguda-Kazimierowicz, E. Theoretical Model of EphA2-Ephrin A1 Inhibition. Molecules 2018, 23, 1688. https://doi.org/10.3390/molecules23071688
Jedwabny W, Lodola A, Dyguda-Kazimierowicz E. Theoretical Model of EphA2-Ephrin A1 Inhibition. Molecules. 2018; 23(7):1688. https://doi.org/10.3390/molecules23071688
Chicago/Turabian StyleJedwabny, Wiktoria, Alessio Lodola, and Edyta Dyguda-Kazimierowicz. 2018. "Theoretical Model of EphA2-Ephrin A1 Inhibition" Molecules 23, no. 7: 1688. https://doi.org/10.3390/molecules23071688
APA StyleJedwabny, W., Lodola, A., & Dyguda-Kazimierowicz, E. (2018). Theoretical Model of EphA2-Ephrin A1 Inhibition. Molecules, 23(7), 1688. https://doi.org/10.3390/molecules23071688