
Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors

Abstract
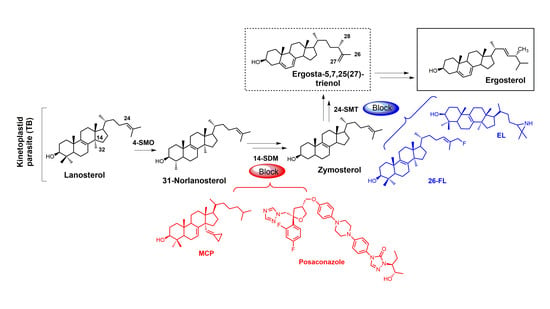
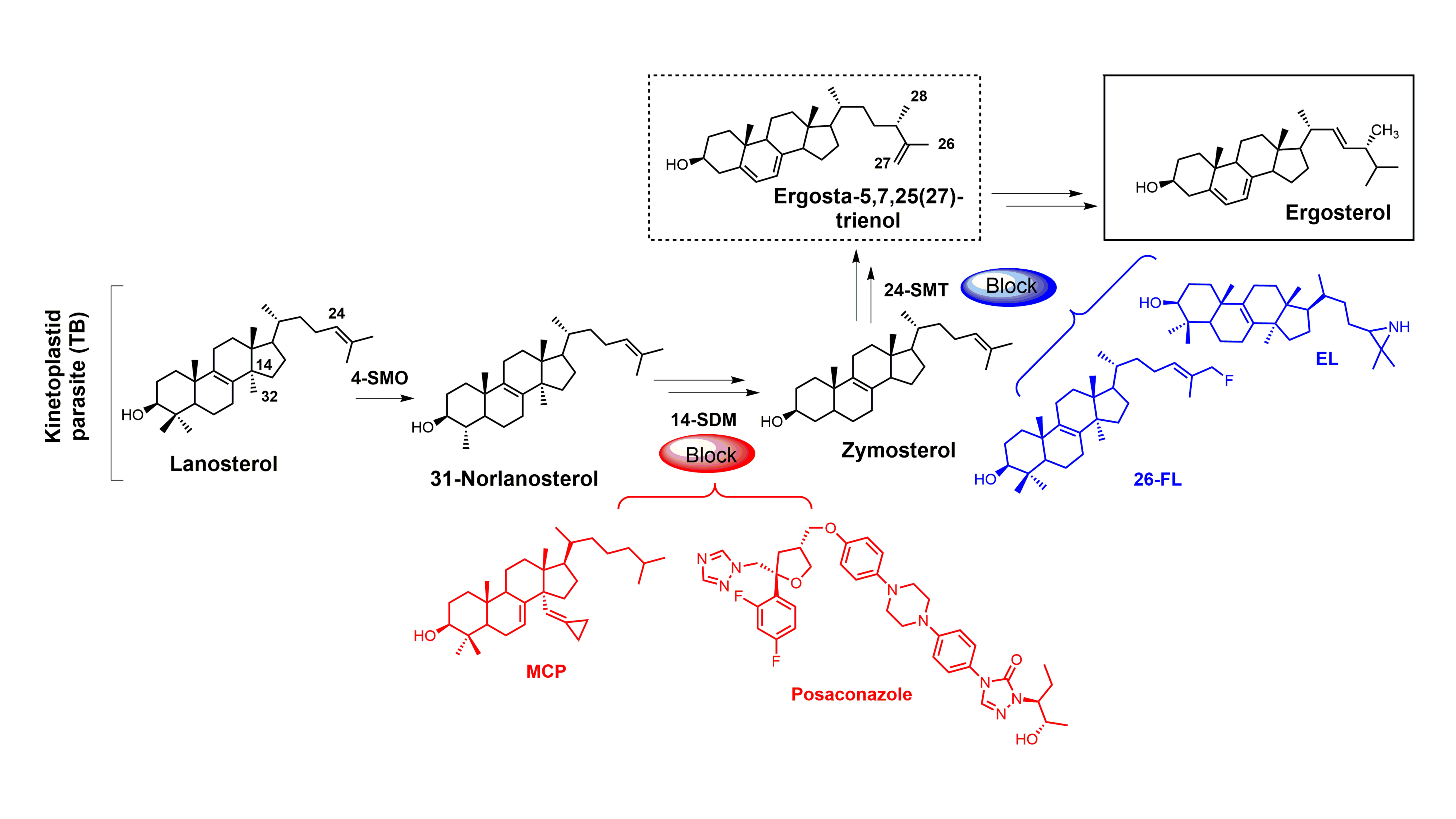
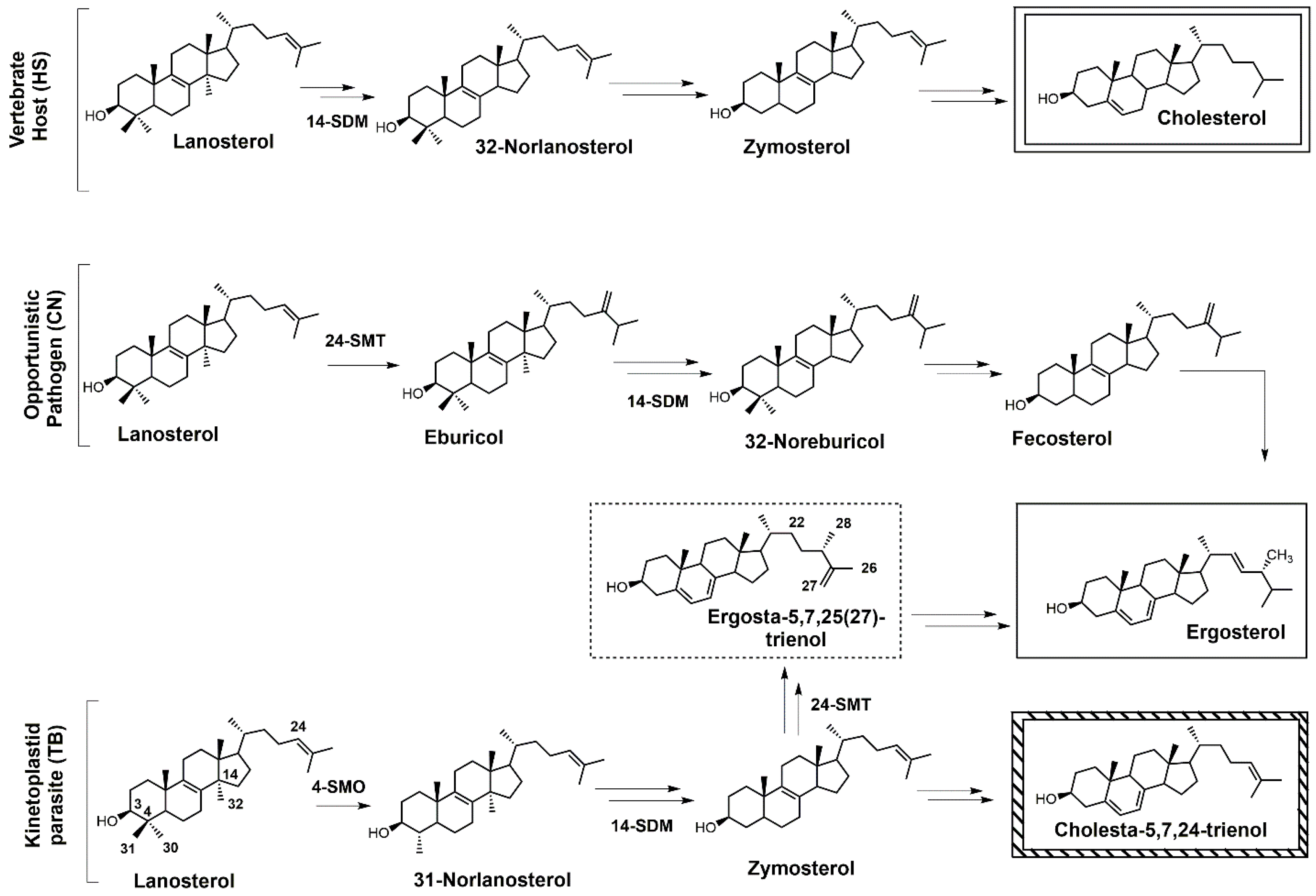
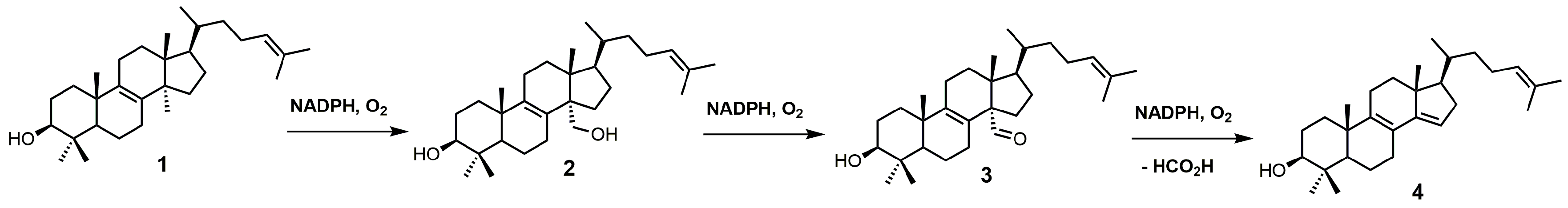
:
{kind=link}
{kind=link}
{kind=link}
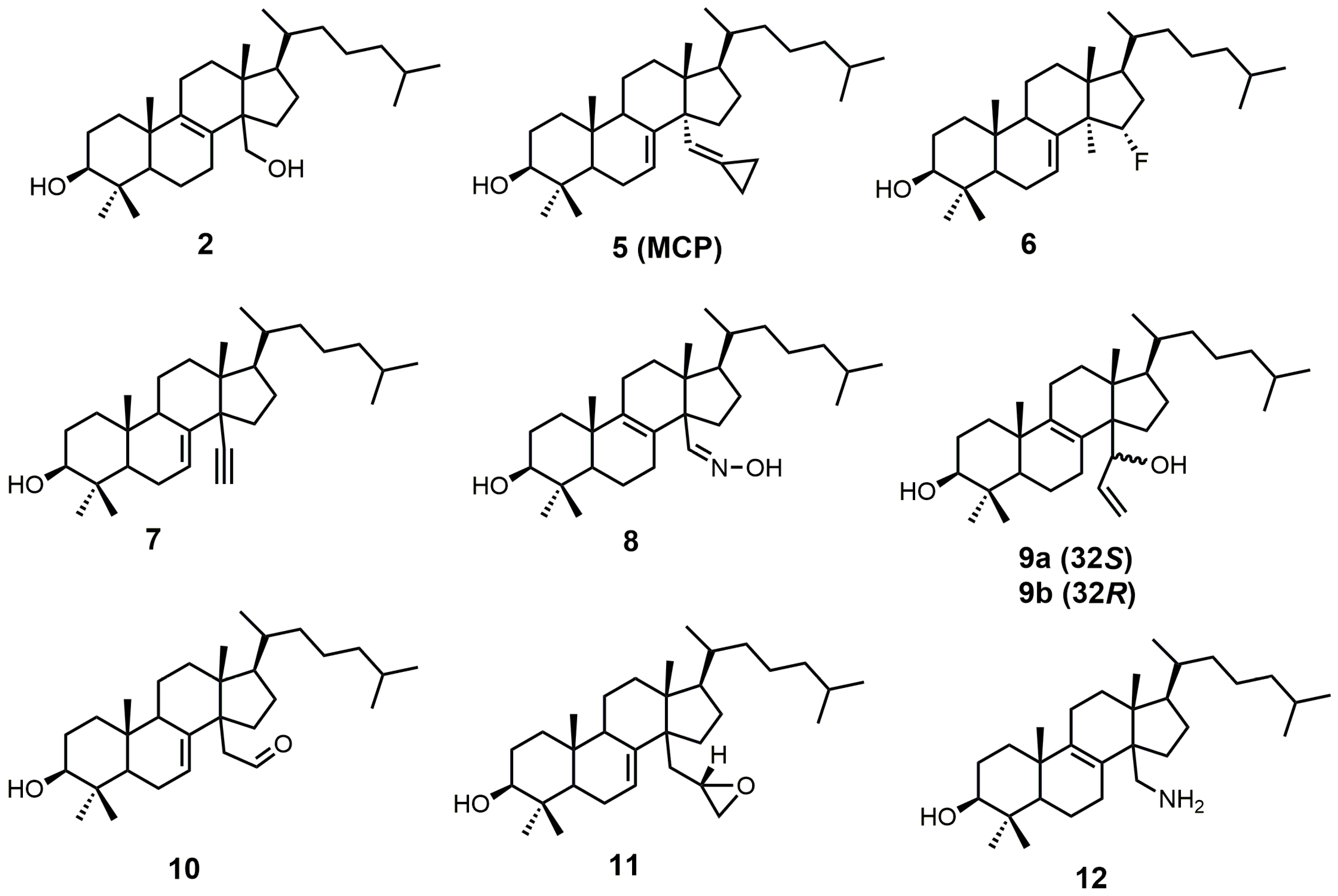{kind=link}
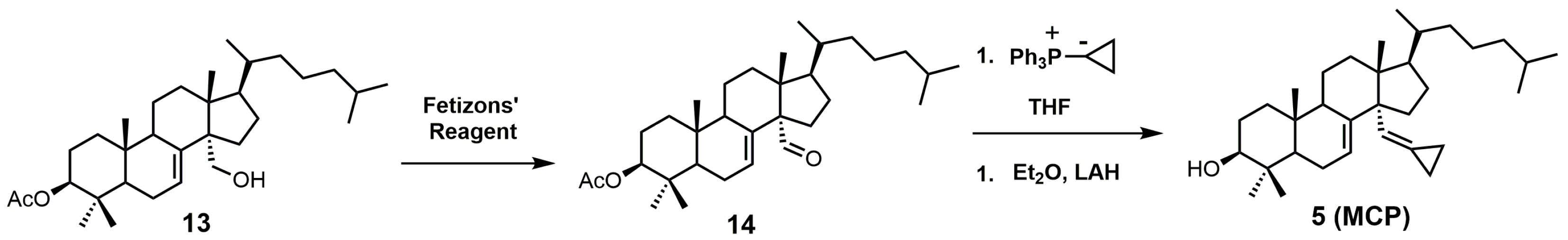{kind=link}
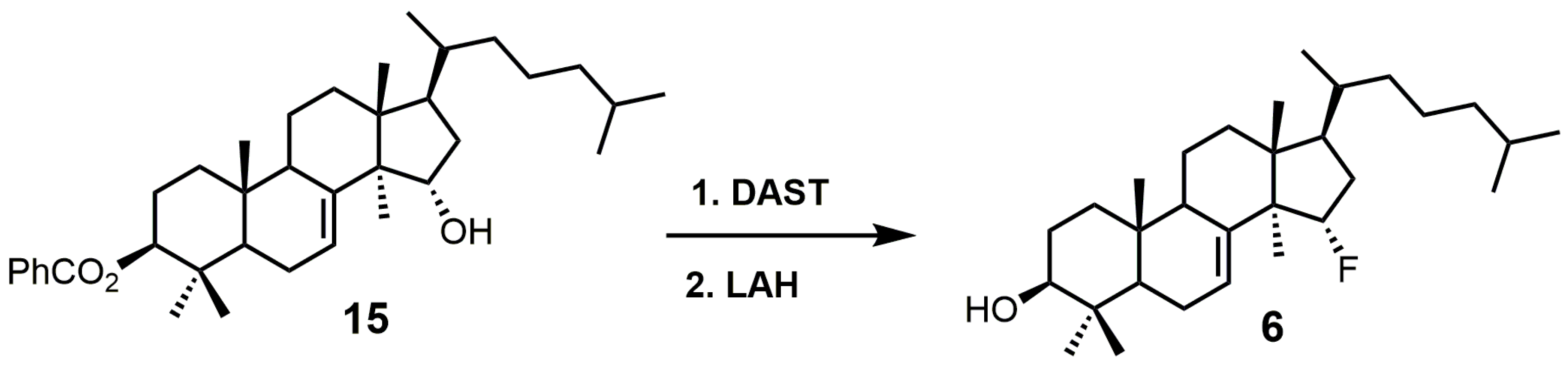{kind=link}
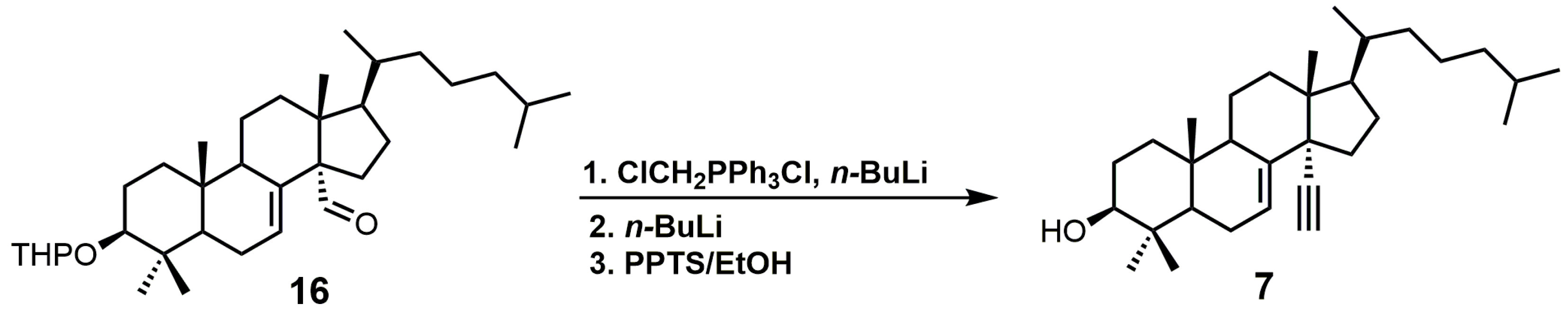{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
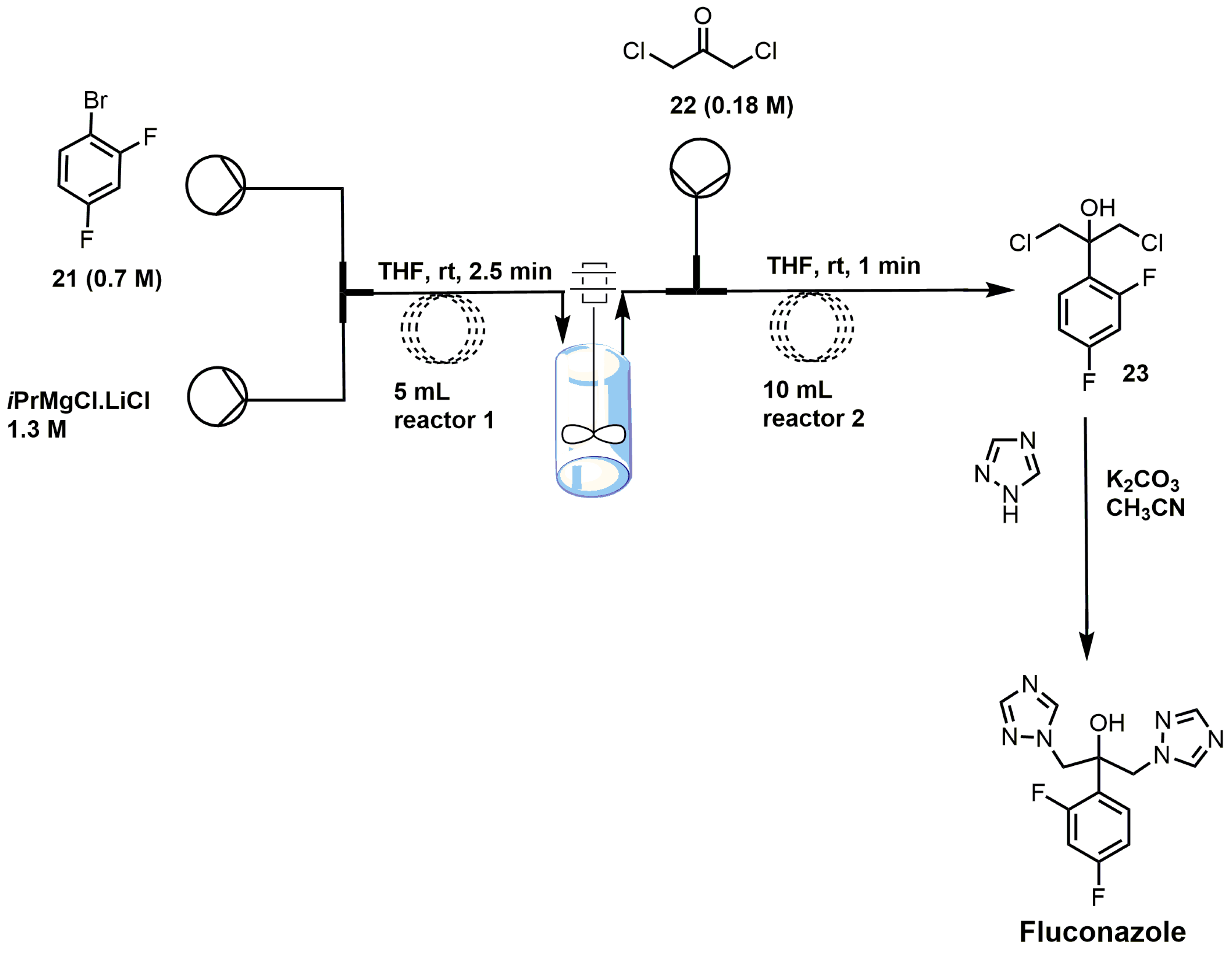{kind=link}
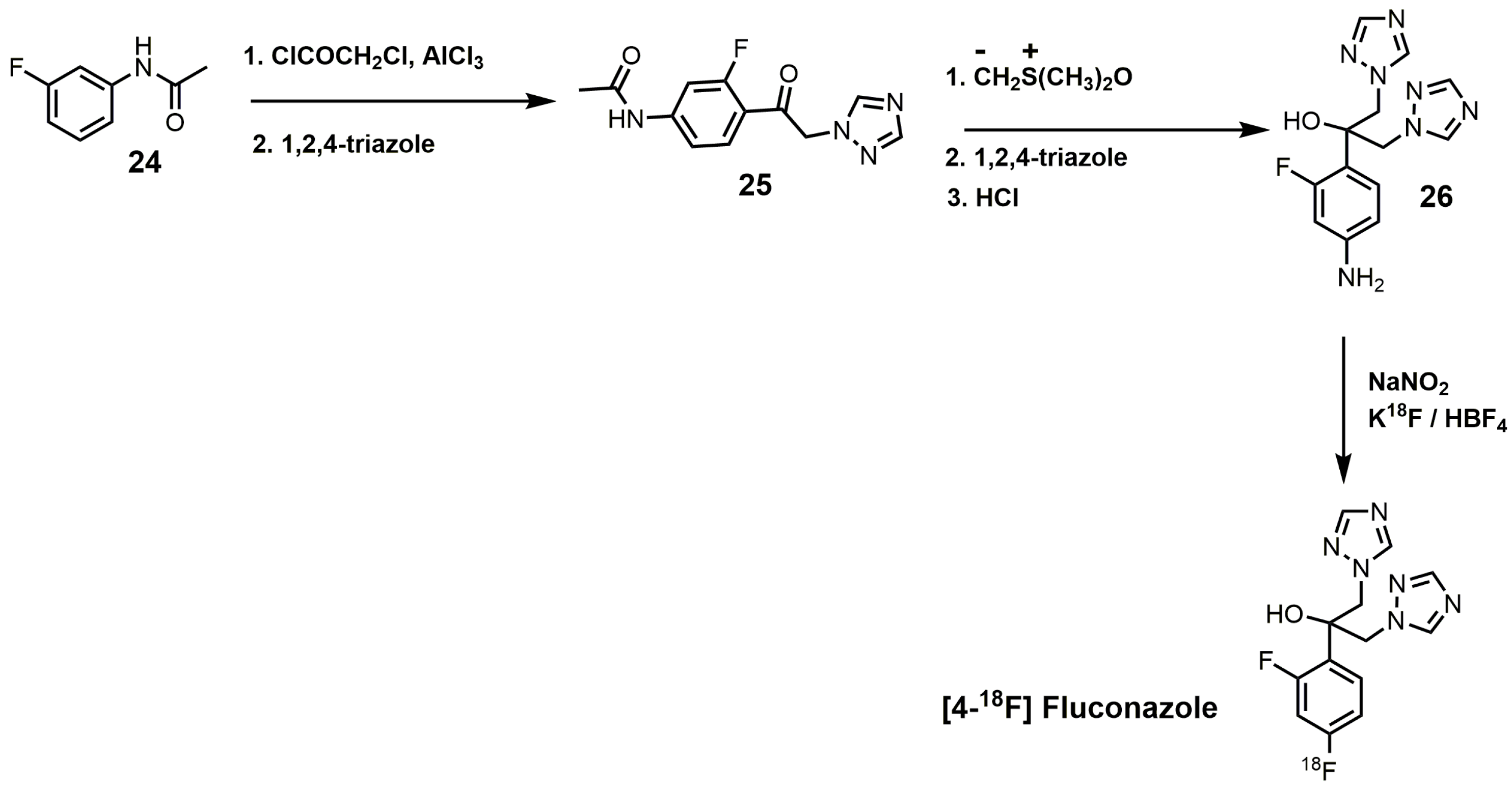{kind=link}
{kind=link}
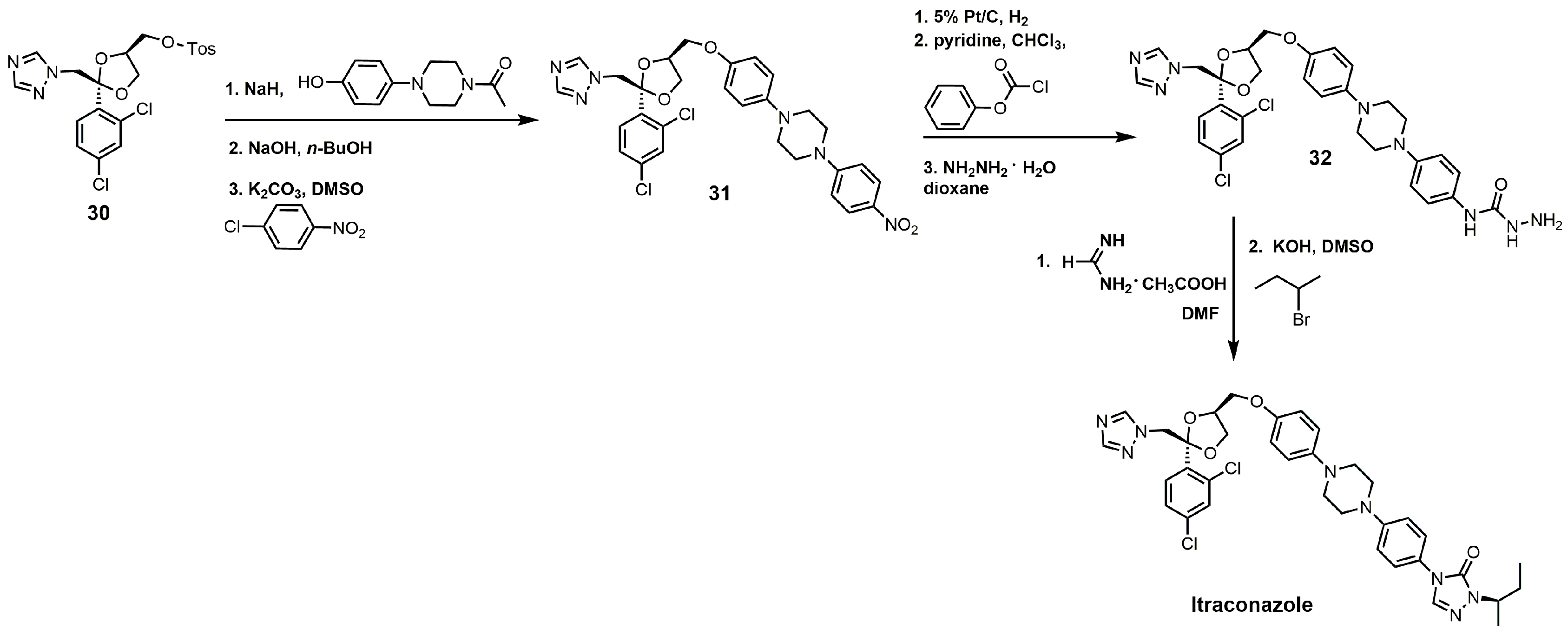{kind=link}
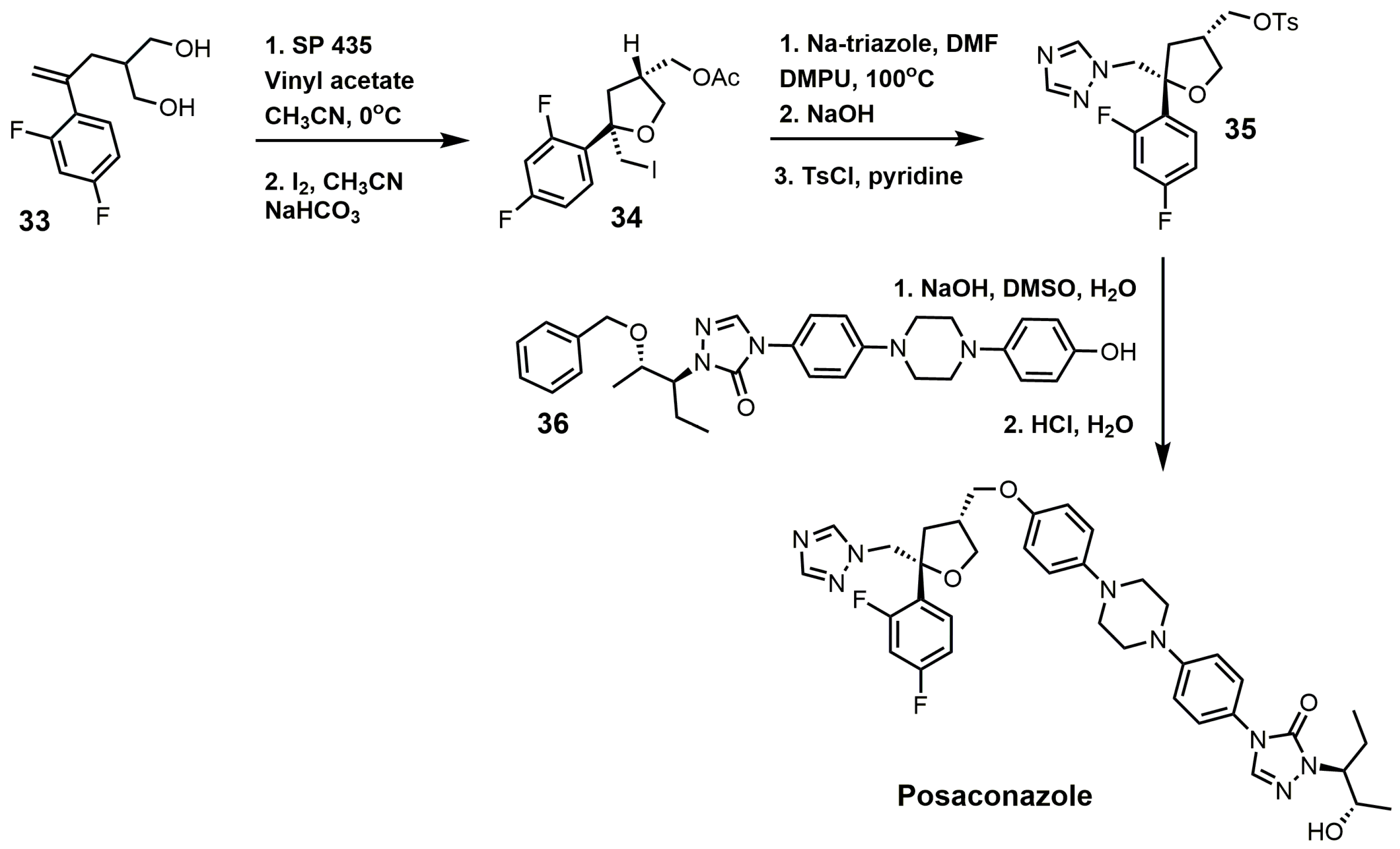{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
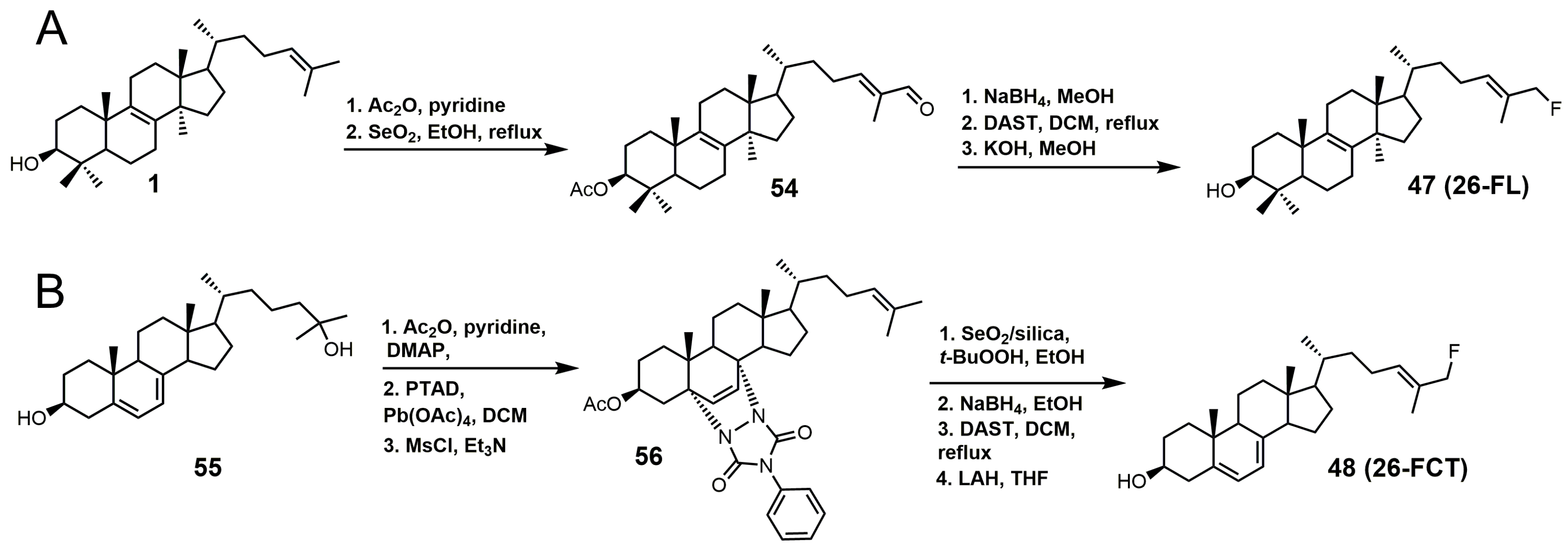{kind=link}
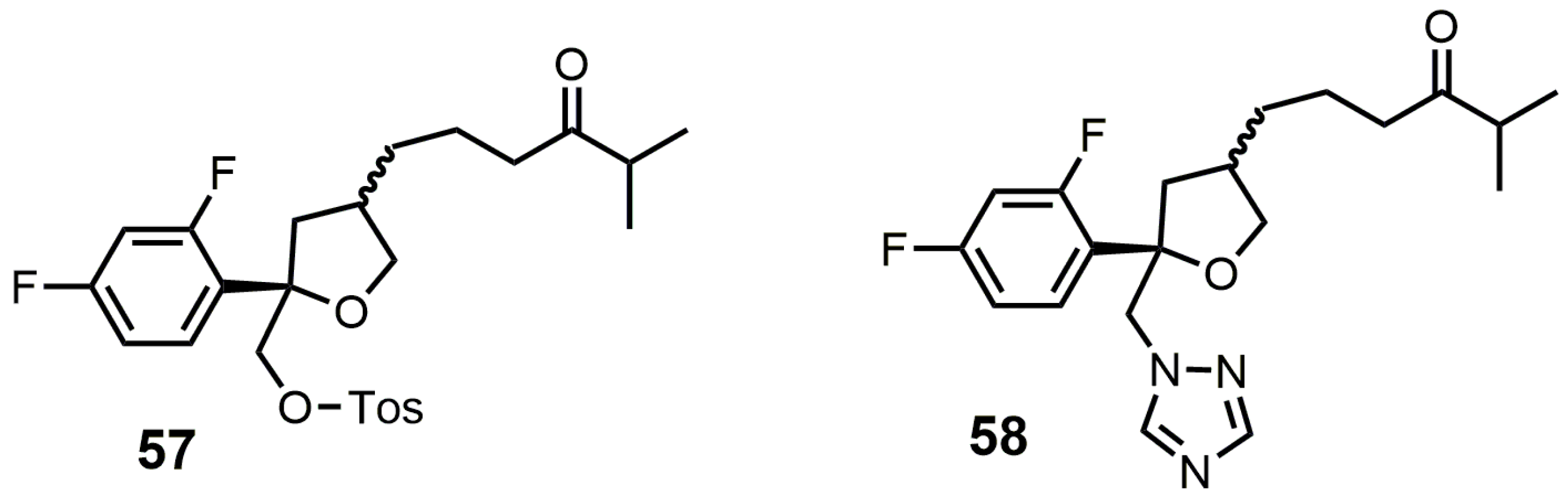{kind=link}
1. Introduction
2. Sterol SDM Inhibitors
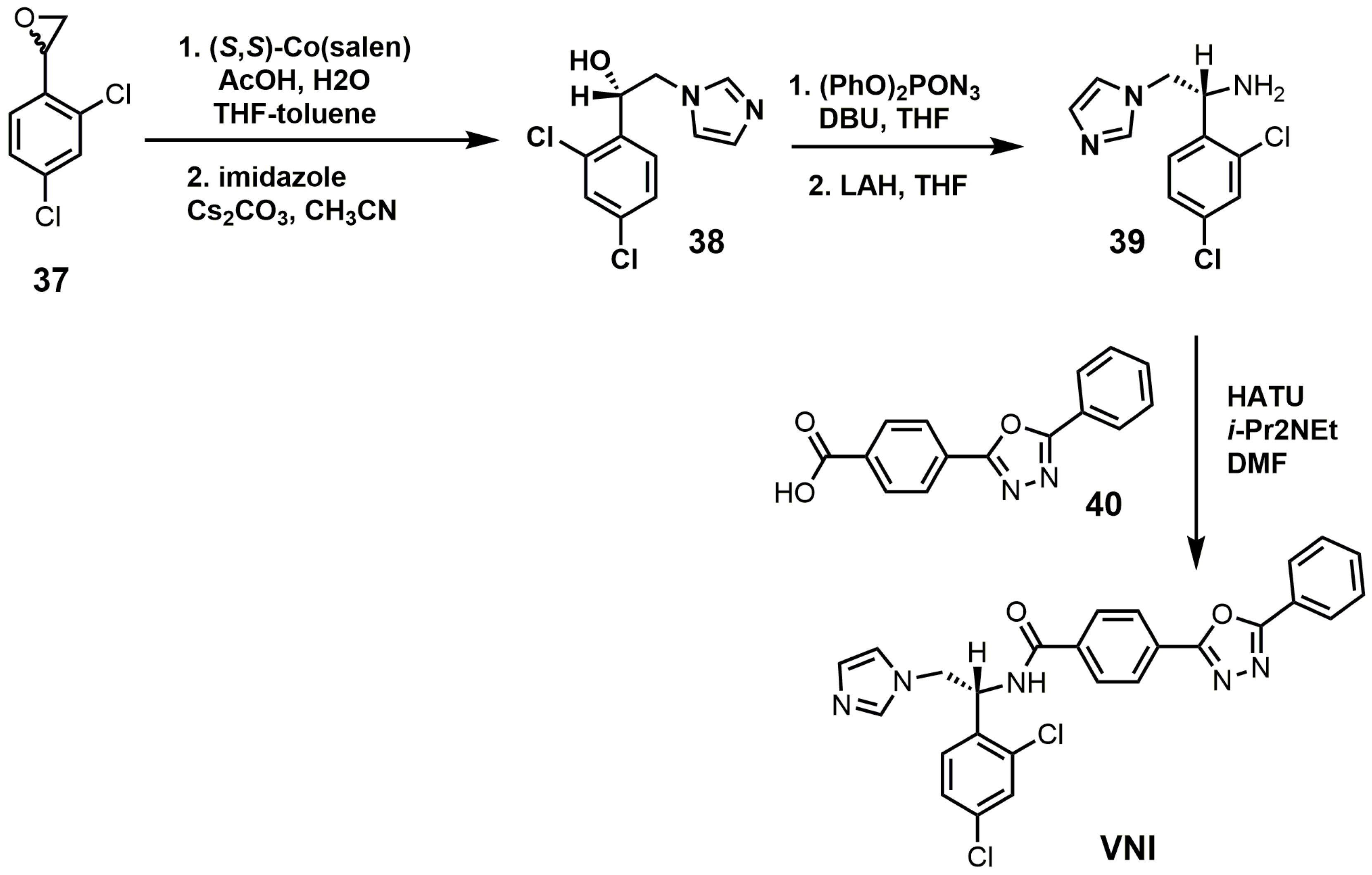
3. Azole SDM Inhibitors
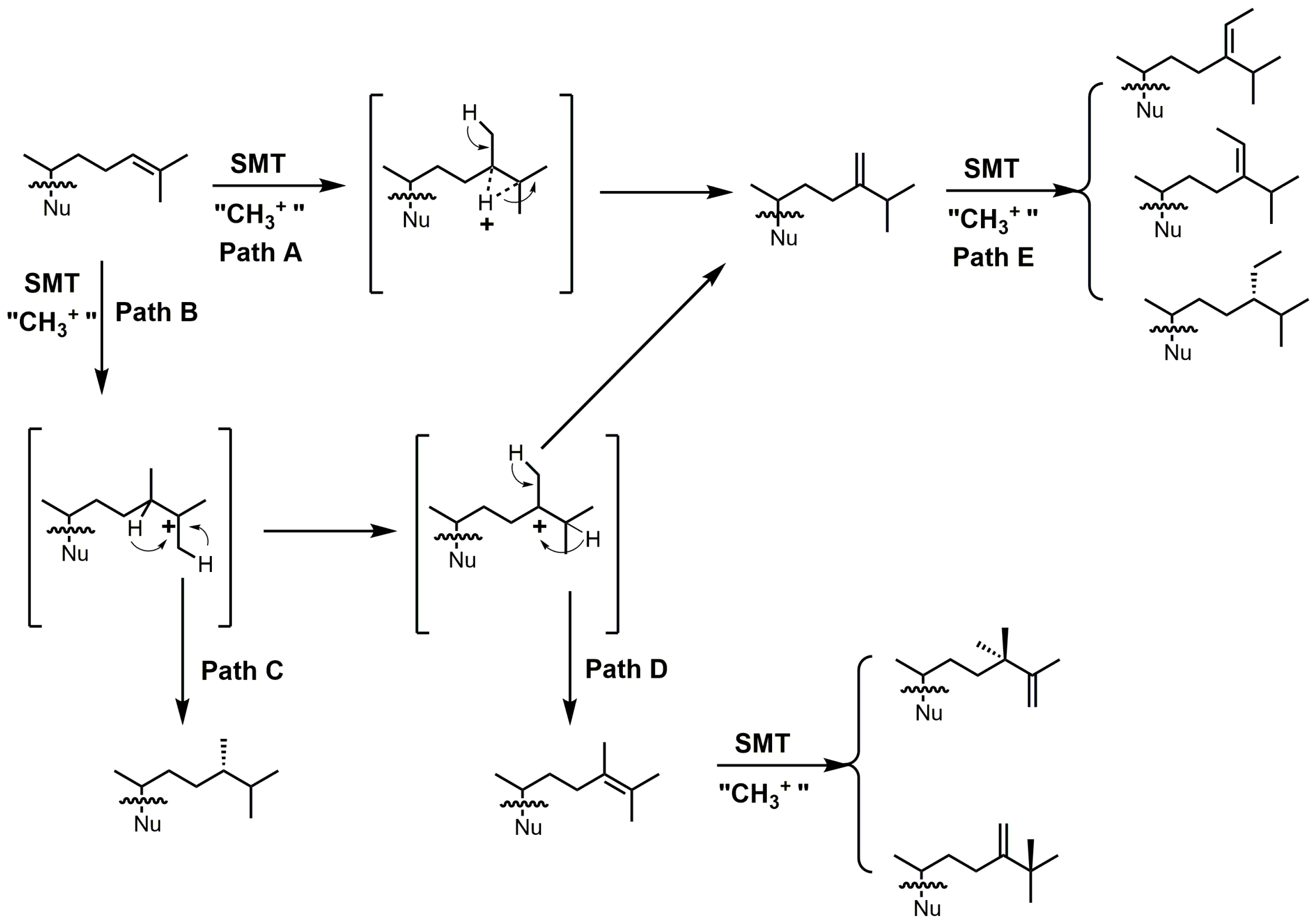
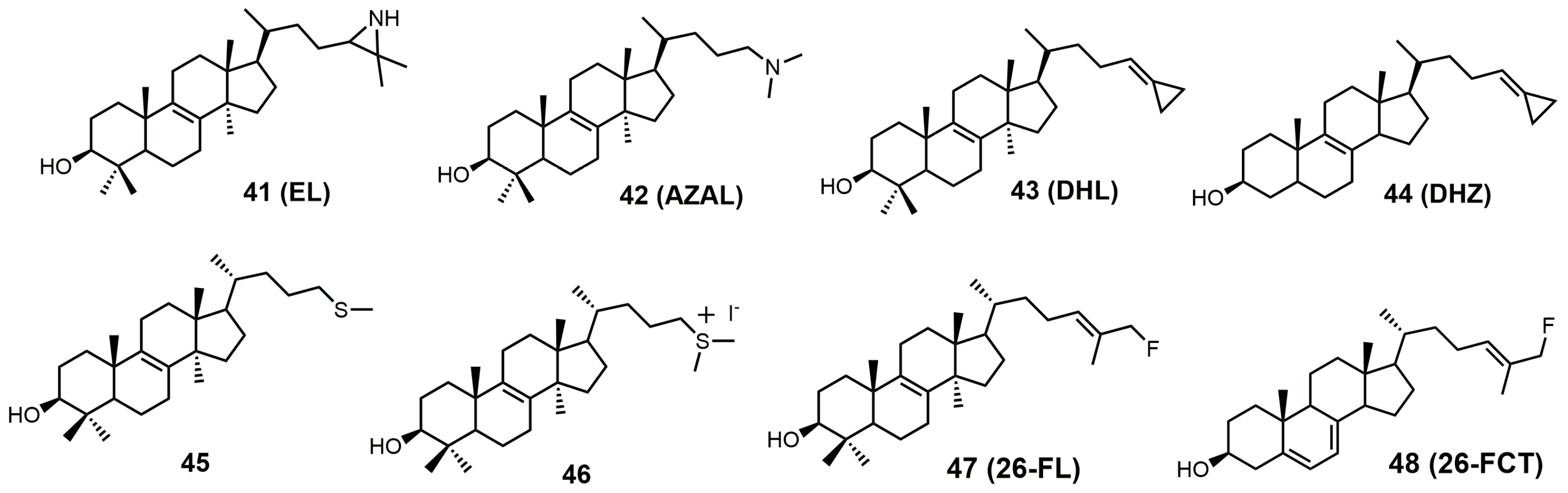
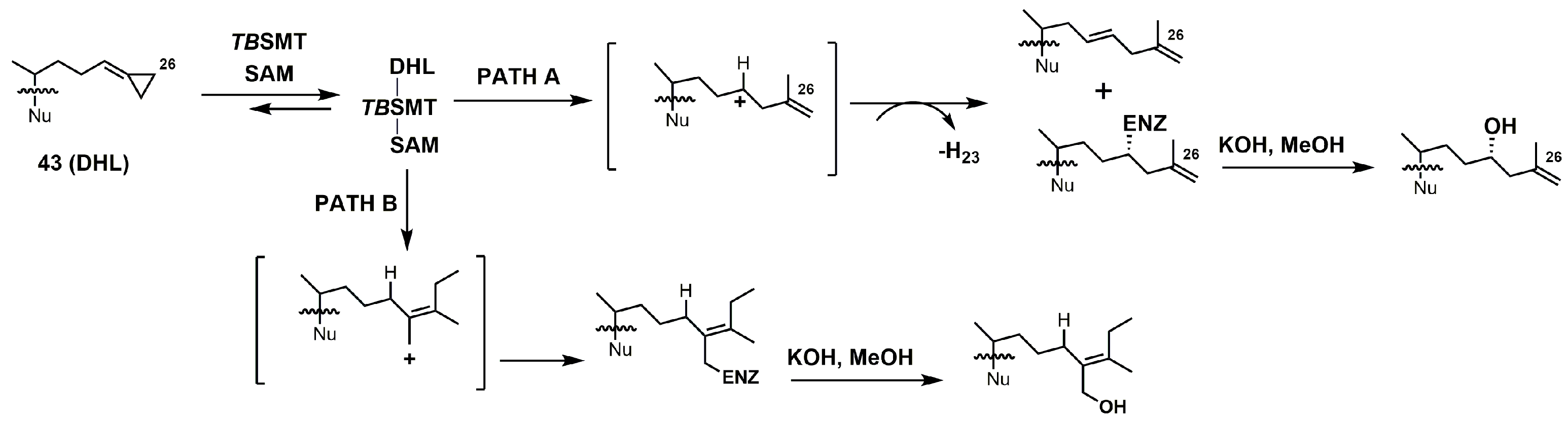
4. 24-SMT Inhibitors
5. Bifunctional 24-SMT and SDM Inhibitors
6. Conclusions
Funding
Conflicts of Interest
References
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef] [PubMed]
- Haubrich, B.A.; Singha, U.K.; Miller, M.B.; Nes, C.R.; Anyatonwu, H.; Lecordier, L.; Patkar, P.; Leaver, D.J.; Villalta, F.; Vanhollebeke, B.; et al. Discovery of an ergosterol-signaling factor that regulates Trypanosoma brucei growth. J. Lipid Res. 2015, 56, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, K.; Kanagasabai, R.; Nguyen, T.T.M.; Nes, W.D. Purification, characterization and inhibition of sterol C24-methyltransferase from Candida albicans. Arch. Biochem. Biophys. 2011, 505, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nes, W.D. Steroidal triterpenes: Design of substrate-based inhibitors of ergosterol and sitosterol synthesis. Molecules 2009, 14, 4690–4706. [Google Scholar] [CrossRef] [PubMed]
- Leaver, D.J.; Patkar, P.; Singha, U.K.; Miller, M.B.; Haubrich, B.A.; Chaudhuri, M.; Nes, W.D. Fluorinated sterols are suicide inhibitors of ergosterol biosynthesis and growth in Trypanosoma brucei. Chem. Biol. 2015, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Nes, W.D.; Zhou, W.; Ganapathy, K.; Liu, J.; Vatsyayan, R.; Chamala, S.; Hernandez, K.; Miranda, M. Sterol 24-C-methyltransferase: An enzymatic target for the disruption of ergosterol biosynthesis and homeostasis in Cryptococcus neoformans. Arch. Biochem. Biophys. 2009, 481, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Rachakonda, G.; Wawrzak, Z.; Pomel, S.; Cojean, S.; Nde, P.N.; Nes, W.D.; Locuson, C.W.; Calcutt, M.W.; et al. VFV as a new effective CYP51 structure-derived drug candidate for chagas disease and visceral leishmaniasis. J. Infect. Dis. 2015, 212, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Zucca, M.; Scutera, S.; Savoia, D. New chemotherapeutic strategies against malaria, leishmaniasis and trypanosomiases. Curr. Med. Chem. 2013, 20, 502–526. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Tavangar, P.; Keighobadi, M. An overview of azoles targeting sterol 14α-demethylase for antileishmanial therapy. Eur. J. Med. Chem. 2017, 135, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Duschank, V.G.; Couto, A.S. Targets and patented drugs for chemotherapy of Chagas disease. In Frontiers in Anti-Infective Drug Discovery; Choudhary, M.I., Ed.; Bentham Science Publishers: Oak Park, IL, USA, 2010; Volume 1, pp. 323–408. [Google Scholar]
- Simarro, P.P.; Cecchi, G.; Franco, J.R.; Paone, M.; Diarra, A.; Ruiz-Postigo, J.A.; Fѐvre, E.M.; Mattioli, R.C.; Jannin, J.G. Estimating and mapping the population at risk of sleeping sickness. PLoS Negl. Trop. Dis. 2012, 6, e1859. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Meerpoel, L.; Lewi, P. Conazoles. Molecules 2010, 15, 4129–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, H.L.; Ernst, E.J.; Klepser, M.E. Novel triazole antifungal agents. Exp. Opin. Investig. Drugs 2000, 9, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Kim, K.; Soeiro, M.N.C.; da Silva, C.F.; Batista, D.G.J.; Batista, M.M.; Yazlovitskaya, E.M.; Waterman, M.R.; Sulikowski, G.A.; Lepesheva, G.I. CYP51 structures and structure-based development of novel, pathogen-specific inhibitory scaffolds. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Song, Z.; Kanagasabai, R.; Liu, J.; Jayasimha, P.; Sinha, A.; Veeramachanemi, P.; Miller, M.B.; Nes, W.D. Mechanism-based enzyme inactivators of phytosterol biosynthesis. Molecules 2004, 9, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [PubMed]
- Worthington, P. Sterol biosynthesis inhibiting triazole fungicides. In Bioactive Heterocyclic Compound Classes: Agrochemicals; Lamberth, C., Dinges, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 129–145. [Google Scholar]
- Parker, J.E.; Warrilow, A.G.S.; Cools, H.J.; Martel, C.M.; Nes, W.D.; Fraaije, B.A.; Lucas, J.A.; Kelly, D.E.; Kelly, S.L. Mechanism of binding of prothioconazole to Mycosphaerella graminicola CYP51 differs from that of other azole antifungals. Appl. Environ. Microbiol. 2011, 77, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Price, C.L.; Warrilow, A.G.S.; Parker, J.E.; Mullins, J.G.L.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. Novel substrate specificity and temperature-sensitive activity of Mycosphaerella graminicola CYP51 supported by the native NADPH cytochrome P450 reductase. Appl. Environ. Microbiol. 2015, 81, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Price, C.L.; Parker, J.E.; Warrilow, A.G.S.; Kelly, D.E.; Kelly, S.L. Azole fungicides–understanding resistance mechanisms in agricultural fungal pathogens. Pest Manag. Sci. 2015, 71, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Anderson, S.; Kleshchenko, Y.; Furtak, V.; Wawrzak, Z.; Villalta, F.; Waterman, M.R. Structural insights into inhibition of sterol 14α-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 2010, 285, 25582–25590. [Google Scholar] [CrossRef] [PubMed]
- Tuck, S.F.; Robinson, C.H.; Silverton, J.V. Assessment of the active-site requirements of lanosterol 14α-demethylase: Evaluation of novel substrate analogues as competitive inhibitors. J. Org. Chem. 1991, 56, 1260–1266. [Google Scholar] [CrossRef]
- Frye, L.L.; Cusack, K.P.; Leonard, D.A. 32-Methyl-32-oxylanosterols: Dual-action inhibitors of cholesterol biosynthesis. J. Med. Chem. 1993, 36, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-7-24,25-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Trzaskos, J.M.; Ko, S.S.; Magolda, R.L.; Favata, M.F.; Fischer, R.T.; Stam, S.H.; Johnson, P.R.; Gaylor, J.L. Substrate-based inhibitors of lanosterol 14α-methyl demethylase: I. Assessment of inhibitor structure-activity relationship and cholesterol biosynthesis inhibition properties. Biochemistry 1995, 34, 9670–9676. [Google Scholar] [CrossRef] [PubMed]
- Trzaskos, J.M.; Fischer, R.T.; Ko, S.S.; Magolda, R.L.; Stam, S.H.; Johnson, P.R.; Gaylor, J.L. Substrate-based inhibitors of lanosterol 14α-methyl demethylase: II. Time-dependent enzyme inactivation by selected oxylanosterol analogs. Biochemistry 1995, 34, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Tuck, S.F.; Patel, H.; Safi, E.; Robinson, C.H. Lanosterol 14α-demethylase (P45014DM): Effects of P45014DM inhibitors on sterol biosynthesis downstream of lanosterol. J. Lipid Res. 1991, 32, 893–902. [Google Scholar] [PubMed]
- Trzaskos, J.M.; Magolda, R.L.; Favata, M.F.; Fischer, R.T.; Johnson, P.R.; Chen, H.W.; Ko, S.S.; Leonard, D.A.; Gaylor, J.L. Modulation of 3-hydroxy-3-methylglutaryl-CoA reductase by 14α-flurolanost-7-en-3-ol. J. Biol. Chem. 1993, 268, 22591–22599. [Google Scholar] [PubMed]
- Frye, L.L.; Robinson, C.H. Synthesis of potential mechanism-based inactivators of lanosterol 14α-methyl demethylase. J. Org. Chem. 1990, 55, 1579–1584. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Zaitseva, N.G.; Nes, W.D.; Zhou, W.; Arase, M.; Liu, J.; Hill, G.C.; Waterman, M.R. CYP51 from Trypanosoma cruzi: A phyla-specific residue in the B’ helix defines substrate preferences of sterol 14α-demethylase. J. Biol. Chem. 2006, 281, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Backx, L.J.J.; Mostmans, J.H.; Van Cutsem, J. Antimycotic imidazoles. Part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. J. Med. Chem. 1979, 22, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Van Cutsem, J. Antimycotic Imidazoles. 5. Synthesis and antimycotic properties of 1-[[2-aryl-4-(arylalkyl)-1,3-dioxolan-3-yl]methyl]-1H-imidazoles. J. Med. Chem. 1981, 24, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Hendrickx, R.; Van Cutsem, J. Antimycotic azoles. 6. Synthesis and antifungal properties of terconazole, a novel triazole ketal. J. Med. Chem. 1983, 26, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Heeres, J.; Backx, L.J.J.; Van Cutsem, J. Antimycotic azoles. 7. Synthesis and antifungal properties of a series of novel triazol-3-ones. J. Med. Chem. 1984, 27, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.; Cooper, K.; Marriott, M.S.; Tarbit, M.H.; Troke, P.F.; Whittle, P.J. Discovery of fluconazole, a novel antifungal agent. Rev. Infect. Dis. 1990, 12, S267–S271. [Google Scholar] [CrossRef] [PubMed]
- Saksena, A.K.; Girijavallabhan, V.M.; Wang, H.; Liu, Y.T.; Pike, R.E.; Ganguly, A.K. Concise asymmetric routes to 2,2,4-trisubstituted tetrahydrofurans via chiral titanium imide enolates: Key intermediates towards synthesis of highly active azole antifungals SCH 51048 and SCH 56592. Tetrahedron Lett. 1996, 37, 5657–5660. [Google Scholar] [CrossRef]
- Saksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Liu, Y.T.; Pinto, P.; Bennett, F.; Jao, E.; Patel, N.; et al. Advances in the chemistry of novel broad-spectrum orally active azole antifungals: Recent studies leading to the discovery of SCH 56592. Spec. Publ. R. Soc. Chem. 1997, 198, 180–199. [Google Scholar] [CrossRef]
- Livni, E.; Fischman, A.J.; Ray, S.I.; Elmaleh, D.R.; Alpert, N.M.; Weiss, S.; Correia, J.A.; Webb, D.; Dahl, R.; Robeson, W.; et al. Synthesis of 18F-labeled fluconazole and positron emission tomography studies in rabbits. Nucl. Med. Biol. 1992, 19, 191–199. [Google Scholar] [CrossRef]
- Korwar, S.; Amir, S.; Tosso, P.N.; Desai, B.K.; Kong, C.J.; Fadnis, S.; Telgang, N.S.; Ahmad, S.; Roper, T.D.; Gupton, B.F. The application of a continuous Grignard reaction in the preparation of fluconazole. Eur. J. Org. Chem. 2017, 6495–6498. [Google Scholar] [CrossRef]
- Aher, N.G.; Pore, V.S.; Mishra, N.N.; Kumar, A.; Shukla, P.K.; Sharma, A.; Bhat, M.K. Synthesis and antifungal activity of 1,2,3-triazole containing fluconazole analogues. Bioorg. Med. Chem. Lett. 2009, 19, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M.; Garvey, E.P.; Shaver, S.R.; Schotzinger, R.J.; Hoekstra, W.J. Design and optimization of highly-selective, broad spectrum fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3243–3248. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Parker, J.E.; Price, C.L.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Wiederhold, N.P.; Nes, W.D.; Kelly, D.E.; Kelly, S.L. The tetrazole VT-1161 is a potent inhibitor of Trichophyton rubrum through its inhibition of T. rubrum CYP51. Antimicrob. Agents Chemother. 2017, 61, e00333-17/1–e00333-12/11. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.S.; Hull, C.M.; Rolley, N.J.; Parker, J.E.; Nes, W.D.; Smith, S.N.; Kelly, D.E.; Kelly, S.L. Clotrimazole as a potent agent for treating the oomycete fish pathogen Saprolegnia parasitica through inhibition of sterol 14α-demethylase (CYP51). Appl. Environ. Microbiol. 2014, 80, 6154–6166. [Google Scholar] [CrossRef] [PubMed]
- Gagnepain, J.; Maity, P.; Lamberth, C.; Cederbaum, F.; Rajan, R.; Jacob, O.; Blum, M.; Bieri, S. Synthesis and fungicidal activity of novel imidazole-based ketene dithioacetals. Bioorg. Med. Chem. 2018, 26, 2009–2016. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Wawrzak, Z.; Blobaum, A.L.; Rachakonda, G.; Lindsley, C.W.; Villalta, F.; Nes, W.D.; Botta, M.; Guengerich, F.P.; et al. Sterol 14α-demethylase structure-based design of VNI ((R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide)) derivatives to target fungal infections: Synthesis, biological evaluation, and crystallographic analysis. J. Med. Chem. 2018, 61, 5679–5691. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Podust, L.M.; Roush, W.R. Drug strategies targeting CYP51 in neglected tropical diseases. Chem. Rev. 2014, 114, 11242–11271. [Google Scholar] [CrossRef] [PubMed]
- Parish, E.J.; Schroepfer, G.J. Sterol synthesis. A simplified method for the synthesis of 32-oxygenated derivatives of 24,25-dihydrolanosterol. J. Lipid Res. 1981, 22, 859–868. [Google Scholar] [PubMed]
- Lepesheva, G.I.; Hargrove, T.Y.; Kleshchenko, Y.; Nes, W.D.; Villalta, F.; Waterman, M.R. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 2008, 43, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.N.; Quiocho, F.A.; Sass, R.L.; Werness, P.; Emery, H.; Knapp, F.F.; Schroepfer, G.J. Sterol biosynthesis: Establishment of the structure of 3-p-bromobenzoyloxy-5α-cholest-8(14)-en-15-ol. Bioorg. Chem. 1976, 5, 1–10. [Google Scholar] [CrossRef]
- Gibbons, G.F.; Ramanada, K. Synthesis and configuration at C-15 of the epimeric 5-lanost-8-en-3,15-diols. J. Chem. Soc. Chem. Commun. 1975, 6, 213–214. [Google Scholar] [CrossRef]
- Gaylor, J.L.; Johnson, P.R.; Ko, S.S.; Magolda, R.L.; Stam, S.H.; Trzaskos, J.M. Steroid Derivatives Useful as Hypocholesterolemics. U.S. Patent 5034548, 20 August 1991. [Google Scholar]
- Musiol, R.; Kowalczyk, W. Azole antimycotics—A highway to new drugs or a dead end? Curr. Med. Chem. 2012, 19, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Villalta, F.; Dobish, M.C.; Nde, P.N.; Kleshchenko, Y.Y.; Hargrove, T.Y.; Johnson, C.A.; Waterman, M.R.; Johnston, J.N.; Lepesheva, G.I. VNI cures acute and chronic experimental Chagas disease. J. Infect. Dis. 2013, 208, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.; Cuenca-Estrella, M.; Monzon, A.; Rodriguez-Tudela, J.L. In-vitro comparative activity of UR-9825, itraconazole and fluconazole against clinical isolates of Candida spp. J. Antimicrob. Chemother. 1999, 44, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.; Brammer, K.W.; Marriott, M.S.; Troke, P.F. Activity of UK-49,858, a bis triazole derivative, against experimental infections with Candida albicans and Trichophyton mentagrophytes. Antimicrob. Agents Chemother. 1985, 27, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Ott, R.D.; Hargrove, T.Y.; Kleshchenko, Y.Y.; Schuster, I.; Nes, W.D.; Hill, G.C.; Villalta, F.; Waterman, M.R. Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: Enzyme inhibition and parasite cell growth. Chem. Biol. 2007, 14, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Bettiol, E.; Samanovic, M.; Murkin, A.S.; Raper, J.; Buckner, F.; Rodriguez, A. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl. Trop. Dis. 2009, 3, e384. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Leung, S.S.F.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Villalta, F.; Waterman, M.R. Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv. Parasitol. 2011, 75, 65–87. [Google Scholar] [PubMed]
- Alrajhi, A.A.; Ibrahim, E.A.; De Vol, E.B.; Khairat, M.; Faris, R.M.; Maguire, J.H. Fluconazole for the treatment of cutaneous leishmaniasis caused by Lesihmania major. N. Engl. J. Med. 2002, 346, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Payares, G.; Molina, J.; Sanoja, C.; Liendo, A.; Lazzardi, K.; Piras, M.M.; Piras, R.; Perez, N.; Wincker, P.; et al. Cure of short- and long term experimental Chagas’ disease using D0870. Science 1996, 273, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Payares, G.; Sanoja, C.; Lira, R.; Romanha, A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents 2003, 21, 27–38. [Google Scholar] [CrossRef]
- Lepesheva, G.I.; Nes, W.D.; Zhou, W.; Hill, G.C.; Waterman, M.R. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 2004, 43, 10789–10799. [Google Scholar] [CrossRef] [PubMed]
- Vivas, J.; Urbina, J.A.; de Souza, W. Ultrastructural alterations in Tyrpanosoma (Schizotrypanum) cruzi induced by Δ24(25) sterol methyl transferase inhibitors and their combinations with ketoconazole. Int. J. Antimicrob. Agents 1996, 7, 235–240. [Google Scholar] [CrossRef]
- Espinel-Ingroff, A.; Shadomy, S.; Gebhart, R.J. In vitro studies with R 51,211 (Itraconazole). Antimicrob. Agents Chemother. 1984, 26, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Borgers, M.; Van de Ven, M.A. Degenerative changes in fungi after itraconazole treatment. Rev. Infect. Dis. 1987, 9, 33–44. [Google Scholar] [CrossRef]
- Lipp, H.-P. Antifungal agents-clinical pharmacokinetics and drug interactions. Mycoses 2008, 51, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Gubbins, P.O. Mould-active azoles: Pharmacokinetics, drug interactions in neutropenic patients. Curr. Opin. Infect. Dis. 2007, 20, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Lamb, D.C.; Kelly, D.E.; Baldwin, B.C.; Kelly, S.L. Differential inhibition of human CYP3A4 and Candida albicans CYP51 with azole antifungal agents. Chem. Biol. Interact. 2000, 125, 165–175. [Google Scholar] [CrossRef]
- Hart, D.T.; Lauwers, W.J.; Willemsens, G.; Bossche, H.V.; Opperdoes, F.R. Perturbation of sterol biosynthesis by itraconazole and ketoconazole in Leishmania mexicana mexicana infected macrophages. Mol. Biochem. Parasitol. 1989, 33, 123–134. [Google Scholar] [CrossRef]
- Momeni, A.Z.; Jalayer, T.; Emamjomeh, M.; Bashardost, N.; Ghassemi, R.L.; Meghdadi, M.; Javadi, A.; Aminjavaheri, M. Treatment of cutaneous leishmaniasis with itraconazole. Randomized double-blind study. Arch. Dermatol. 1996, 132, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Baroni, A.; Aiello, F.S.; Vozza, A.; Vozza, G.; Faccenda, F.; Brasiello, M.; Ruocco, E. Cutaneous leishmaniasis treated with itraconazole. Dermatol. Ther. 2009, 22 (Suppl. 1), S27–S29. [Google Scholar] [CrossRef] [PubMed]
- Minodier, P.; Parola, P. Cutaneous leishmaniasis treatment. Travel Med. Infect. Dis. 2007, 5, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Chemotherapy of Chagas disease. Curr. Pharm. Des. 2002, 8, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A. Ergosterol biosynthesis for the specific treatment of Chagas disease: From basic science to clinical trials. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery, 1st ed.; Jäger, T., Koch, O., Flohé, L., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 489–514. [Google Scholar]
- Benaim, G.; Sanders, J.M.; Garcia-Marchán, Y.; Colina, C.; Lira, R.; Caldera, A.R.; Payares, G.; Sanoja, C.; Burgos, J.M.; Leo-Rossell, A.; et al. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J. Med. Chem. 2006, 49, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Pinazo, M.; Espinosa, G.; Gállego, M.; López-Chejade, P.L.; Urbina, J.A.; Gascón, J. Case report: Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am. J. Trop. Med. Hyg. 2010, 82, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Saksena, A.K.; Girijavallabhan, V.M.; Lovey, R.G.; Pike, R.E.; Wang, H.; Ganguly, A.K.; Morgan, B.; Zaks, A.; Puar, M.S. Highly stereoselective access to novel 2,2,4-trisubstituted tetrahydrofurans by halocyclization: Practical chemoenzymatic synthesis of SCH 51048, a broad-spectrum orally active antifungal agent. Tetrahedron Lett. 1995, 36, 1787–1790. [Google Scholar] [CrossRef]
- Chidambaram, V.S.; Miryala, A.K.; Wadhwa, L. Process for Preparing Posaconazole and Intermediates Thereof. PCT International Application WO2009141837A2, 26 November 2009. [Google Scholar]
- Charyulu, P.V.R.; Gowda, D.J.C.; Rajmahendra, S.; Raman, M. Crystalline Forms of Posaconazole Intermediate and Process for the Preparation of Amorphous Posaconazole. U.S. PCT International Application WO2017051342A1, 30 March 2017. [Google Scholar]
- Lepesheva, G.I.; Park, H.; Hargrove, T.Y.; Vanhollebeke, B.; Wawrzak, Z.; Harp, J.M.; Sundaramoorthy, M.; Nes, W.D.; Pays, E.; Chaudhuri, M.; et al. Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010, 285, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.; Christov, P.; Sulikowski, G.A.; Kim, K. A convergent, scalable and stereoselective synthesis of azole CYP51 inhibitors. Tetrahedron Lett. 2017, 58, 4248–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nes, W.D.; Janssen, G.G.; Norton, R.A.; Kalinowska, M.; Crumley, F.G.; Tal, B.; Bergenstrahle, A.; Jonsson, L. Regulation of sterol biosynthesis in sunflower by 24(R,S)25-epiminolanosterol, a novel C-24 methyl transferase inhibitor. Biochem. Biophys. Res. Commun. 1991, 177, 566–574. [Google Scholar] [CrossRef]
- Nes, W.D.; Xu, S.; Parish, E.J. Metabolism of 24(R,S),25-epiminolanosterol to 25-aminolanosterol and lanosterol by Gibberella fujikuroi. Arch. Biochem. Biophys. 1989, 272, 323–331. [Google Scholar] [CrossRef]
- Popjak, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of cholesterol synthesis and cell growth by 24(R,S),25-iminolanosterol and triparanol in cultured rat hepatoma cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [PubMed]
- Zhou, W.; Lepesheva, G.I.; Waterman, M.R.; Nes, W.D. Mechanistic analysis of a multiple product sterol methyltransferase implicated in ergosterol biosynthesis in Trypanosoma brucei. J. Biol. Chem. 2006, 281, 6290–6296. [Google Scholar] [CrossRef] [PubMed]
- Urbina, J.A.; Vivas, J.; Lazardi, K.; Molina, J.; Payares, G.; Piras, M.M.; Piras, R. Antiproliferative effects of Δ24(25) sterol methyl transferase inhibitors on Trypanosoma (Schizotrypanum) cruzi: In vitro and in vivo studies. Chemotherapy 1996, 42, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Kidane, M.E.; Vanderloop, B.H.; Zhou, W.; Thomas, C.D.; Ramos, E.; Singha, U.; Chaudhuri, M.; Nes, W.D. Sterol methyltransferase a target for anti-amoebatherapy: Towards transition state analog and suicide substrate drug design. J. Lipid Res. 2017, 58, 2310–2323. [Google Scholar] [CrossRef] [PubMed]
- Parish, E.J.; Nes, W.D. Synthesis of new epiminoisopentenoids. Synth. Commun. 1998, 18, 221–226. [Google Scholar] [CrossRef]
- Nes, W.D. Sterol methyl transferase: Enzymology and inhibition. Biochim. Biophys. Acta 2000, 1529, 63–88. [Google Scholar] [CrossRef]
- Pereira, M.; Song, Z.; Santos-Silva, L.K.; Richards, M.H.; Nguyen, T.T.M.; Liu, J.; Soares, C.M.A.; Cruz, A.H.S.; Ganapathy, K.; Nes, W.D. Cloning, mechanistic and functional analysis of a fungal sterol C24-methyltransferase implicated in brassicasterol biosynthesis. Biochim. Biophys. Acta 2010, 1801, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cross, G.A.M.; Nes, W.D. Cholesterol import fails to prevent catalyst-based inhibition of ergosterol synthesis and cell proliferation of Trypanosoma brucei. J. Lipid Res. 2007, 48, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Kohen, F.; Counsell, R.E. Hypocholesterolemic agents. 8. Synthesis of 25-azadihydrolanosterol and derivatives. J. Med. Chem. 1971, 14, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Oehlschlager, A.C.; Angus, R.H.; Pierce, A.M.; Pierce, H.D.; Srinivasan, R.J. Azasterol inhibition of 24-sterol methyltransferase in Saccharomyces cerevisiae. Biochemistry 1984, 23, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Patkar, P.; Singha, U.K.; Chaudhuri, M.; Nes, W.D. 24-Methylenecyclopropane steroidal inhibitors: A Trojan horse in ergosterol biosynthesis that prevents growth of Trypanosoma brucei. Biochim. Biophys. Acta 2017, 1862, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Nes, W.D. Isolation and characterization of an active-site peptide from a sterol methyl transferase with a mechanism-based inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 1533–1536. [Google Scholar] [CrossRef]
- Neelakandan, A.K.; Song, Z.; Wang, J.; Richards, M.H.; Wu, X.; Valliyodan, B.; Nguyen, H.T.; Nes, W.D. Cloning, functional expression and phylogenetic analysis of plant sterol 24C-methyltransferases involved in sitosterol biosynthesis. Phytochemistry 2009, 70, 1982–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Nes, W.D. Sterol methyltransferase2: Purification, properties, and inhibition. Arch. Biochem. Biophys. 2003, 420, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Zhou, W.; Liu, J.; Nes, W.D. Mechanism-based active site modification of the soybean sterol methyltransferase by 26,27-dehydrocycloartenol. Bioorg. Med. Chem. Lett. 2004, 14, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhou, W.; Guo, D.; Nes, W.D. Synthesis of rationally designed mechanism-based inactivators of the (S)-adenosyl-L-methionine: Δ24(25)-sterol methyl transferase. Synth. Commun. 1996, 26, 3841–3848. [Google Scholar] [CrossRef]
- Kanagasabai, R.; Zhou, W.; Liu, J.; Nguyen, T.T.M.; Veeramachaneni, P.; Nes, W.D. Disruption of ergosterol biosynthesis, growth, and the morphological transition in Candida albicans by sterol methyltransferase inhibitors containing sulfur at C-25 in the sterol side chain. Lipids 2004, 39, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Patkar, P.; Haubrich, B.A.; Qi, M.; Nguyen, T.T.M.; Thomas, C.D.; Nes, W.D. C24-Methylation of 26-fluorocycloartenols by recombinant sterol C24-methyltransferase from soybean: Evidence for channel switching and its phylogenetic implications. Biochem. J. 2013, 456, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Fuse, S.; Mifune, Y.; Tanabe, N.; Takahashi, T. Continuous-flow synthesis of activated vitamin D3 and its analogues. Org. Biomol. Chem. 2012, 10, 5205–5211. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-K.; Lee, K.-W.; Kang, H.I.; Yamashita, C.; Kudo, M.; Yoshida, Y. Design and synthesis of potential inhibitors of the ergosterol biosynthesis as antifungal agents. Bioorg. Med. Chem. 2000, 8, 2475–2486. [Google Scholar] [CrossRef]
- Yang, G.; Lee, N.; Ioset, J.-R.; No, J.H. Evaluation of parameters impacting drug susceptibility in intracellular Trypanosoma cruzi assay protocols. SLAS Discov. 2017, 22, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Prat, J.G.; Salvador, F.; Trevino, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Waskin, H.; Sosa-Estani, S.; del Carmen Bangher, M.; Cuneo, C.; Milesi, R.; Mallagray, M.; Apt, W.; Beloscar, J.; Gascon, J.; et al. Benznidazole and posaconazole in eliminating parasites in asymptomatic T. cruzi carriers: The stop-Chagas trial. J. Am. Coll. Cardiol. 2017, 69, 939–947. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





























© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leaver, D.J. Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors. Molecules 2018, 23, 1753. https://doi.org/10.3390/molecules23071753
Leaver DJ. Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors. Molecules. 2018; 23(7):1753. https://doi.org/10.3390/molecules23071753
Chicago/Turabian StyleLeaver, David J. 2018. "Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors" Molecules 23, no. 7: 1753. https://doi.org/10.3390/molecules23071753
APA StyleLeaver, D. J. (2018). Synthesis and Biological Activity of Sterol 14α-Demethylase and Sterol C24-Methyltransferase Inhibitors. Molecules, 23(7), 1753. https://doi.org/10.3390/molecules23071753