A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels–Alder and Polar Alder-ene Reactions




Abstract
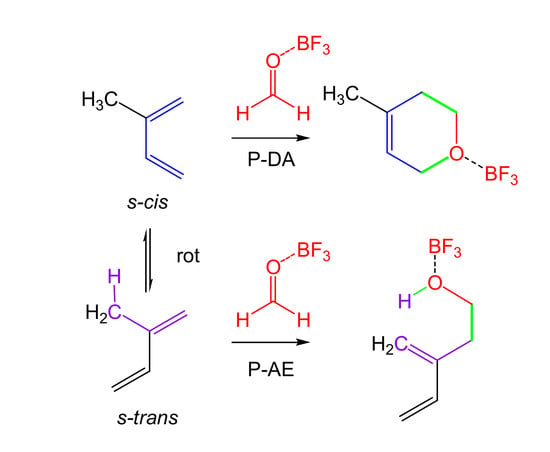
:
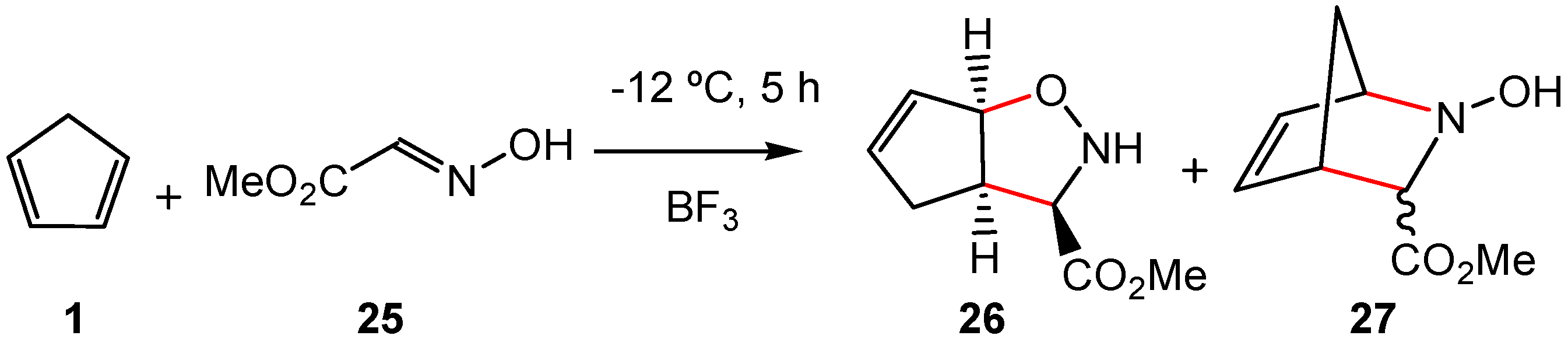
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Analysis of the CDFT Reactivity Indices at the GS of the Reagents
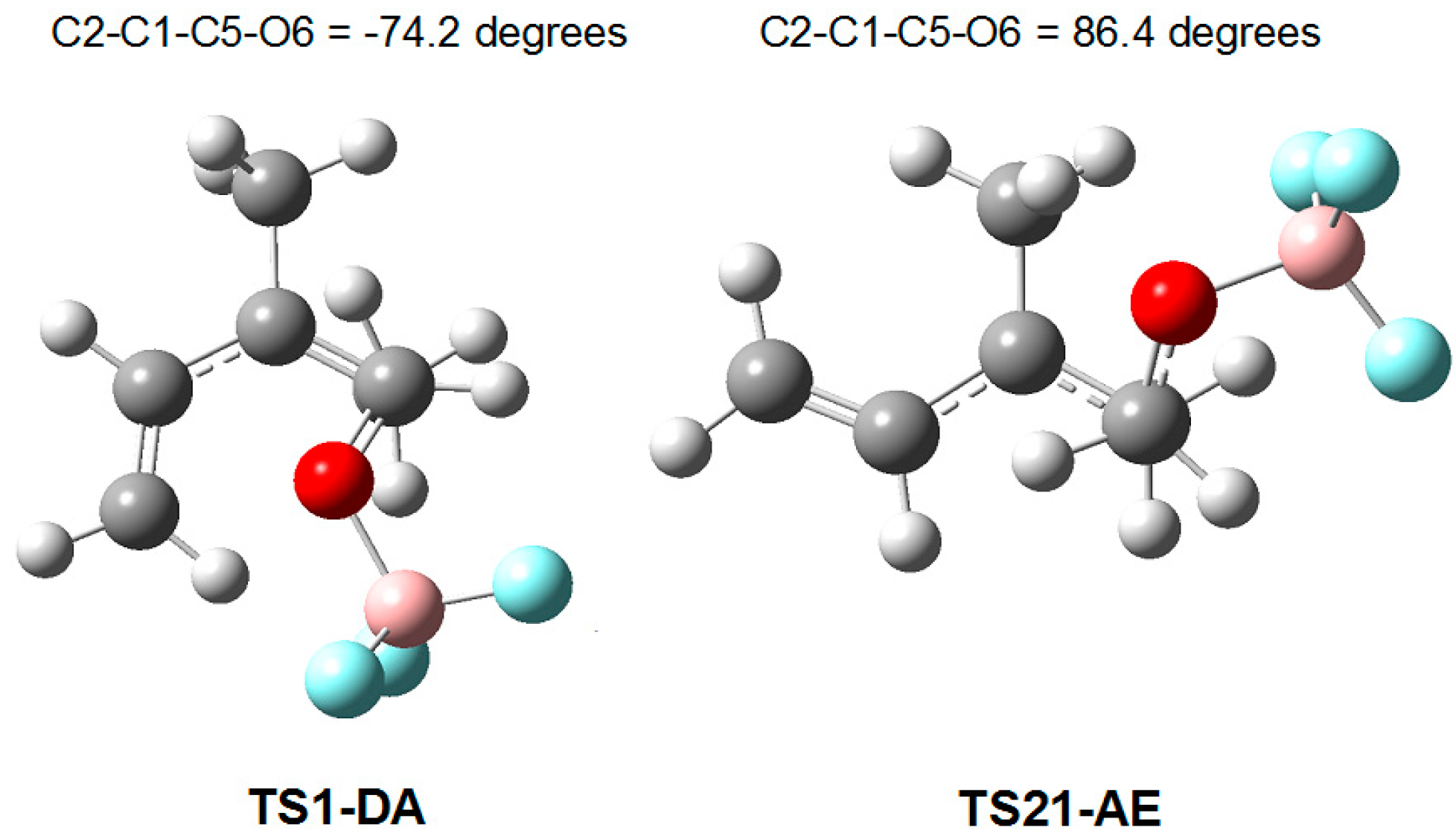
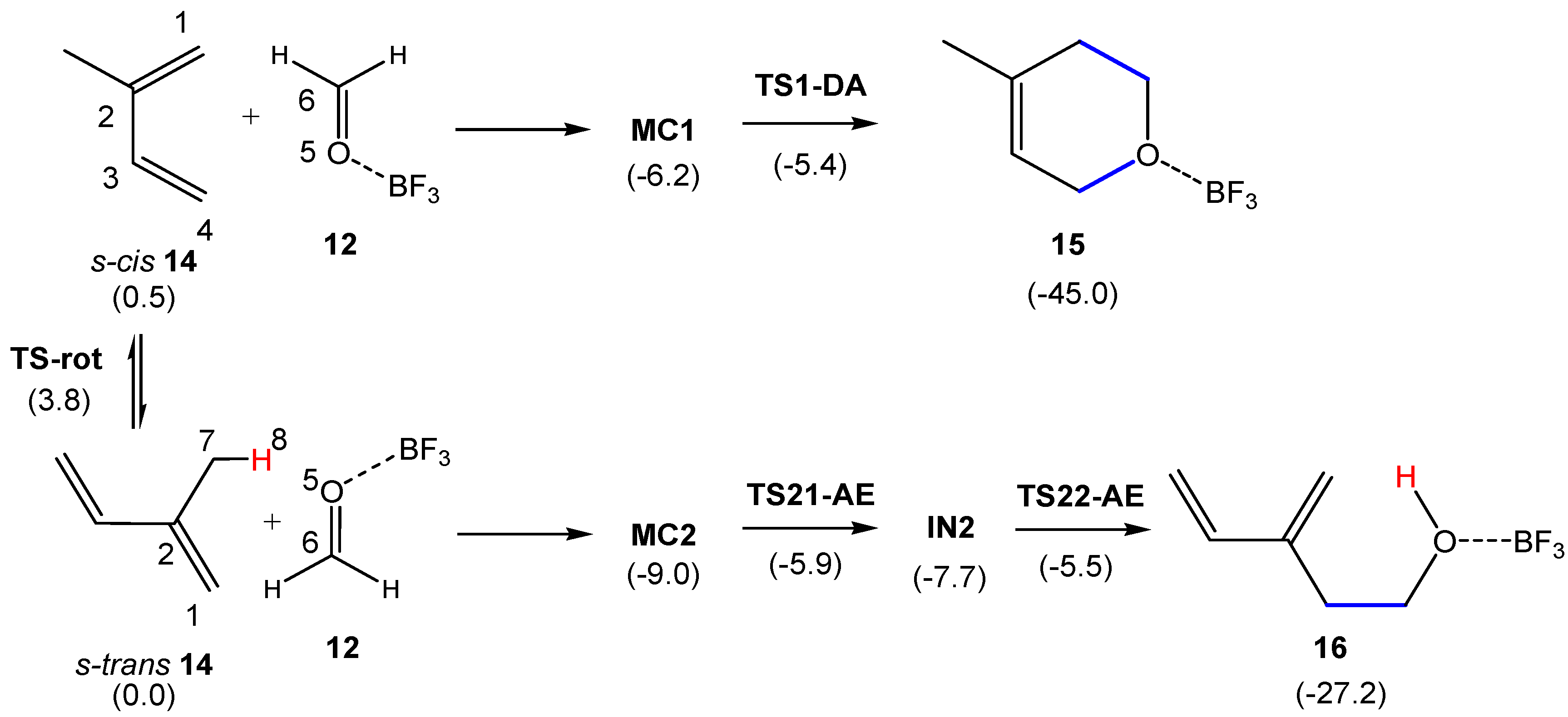
3.2. Comparative Study of the Competitive P-DA and P-AE Reactions between 2MBD 14 and LA Complex 12
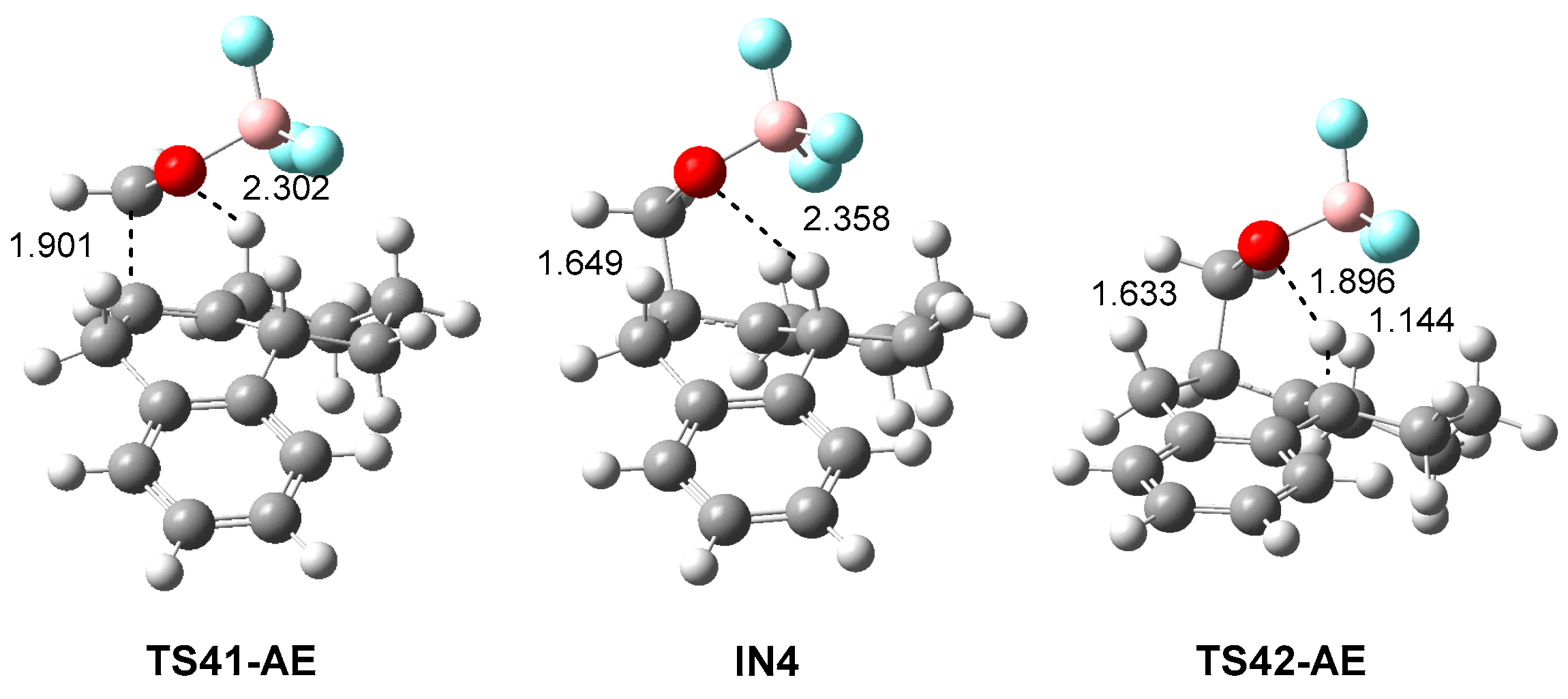
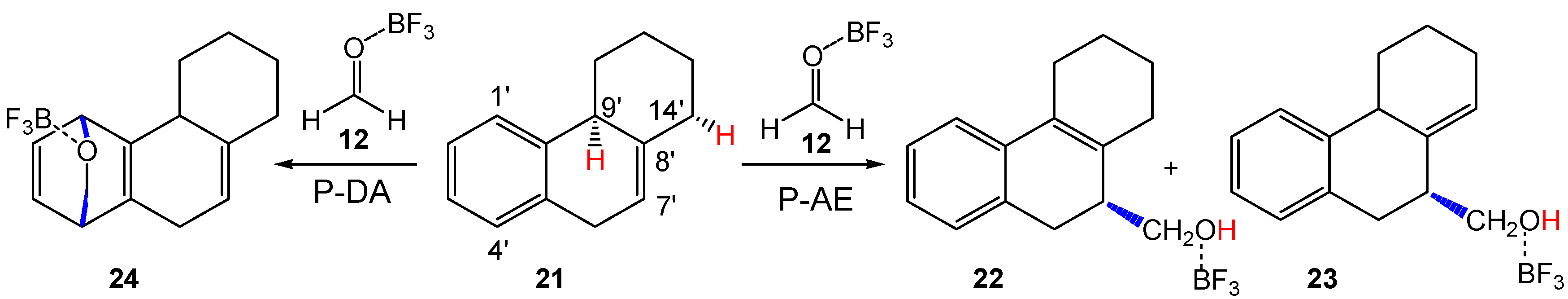
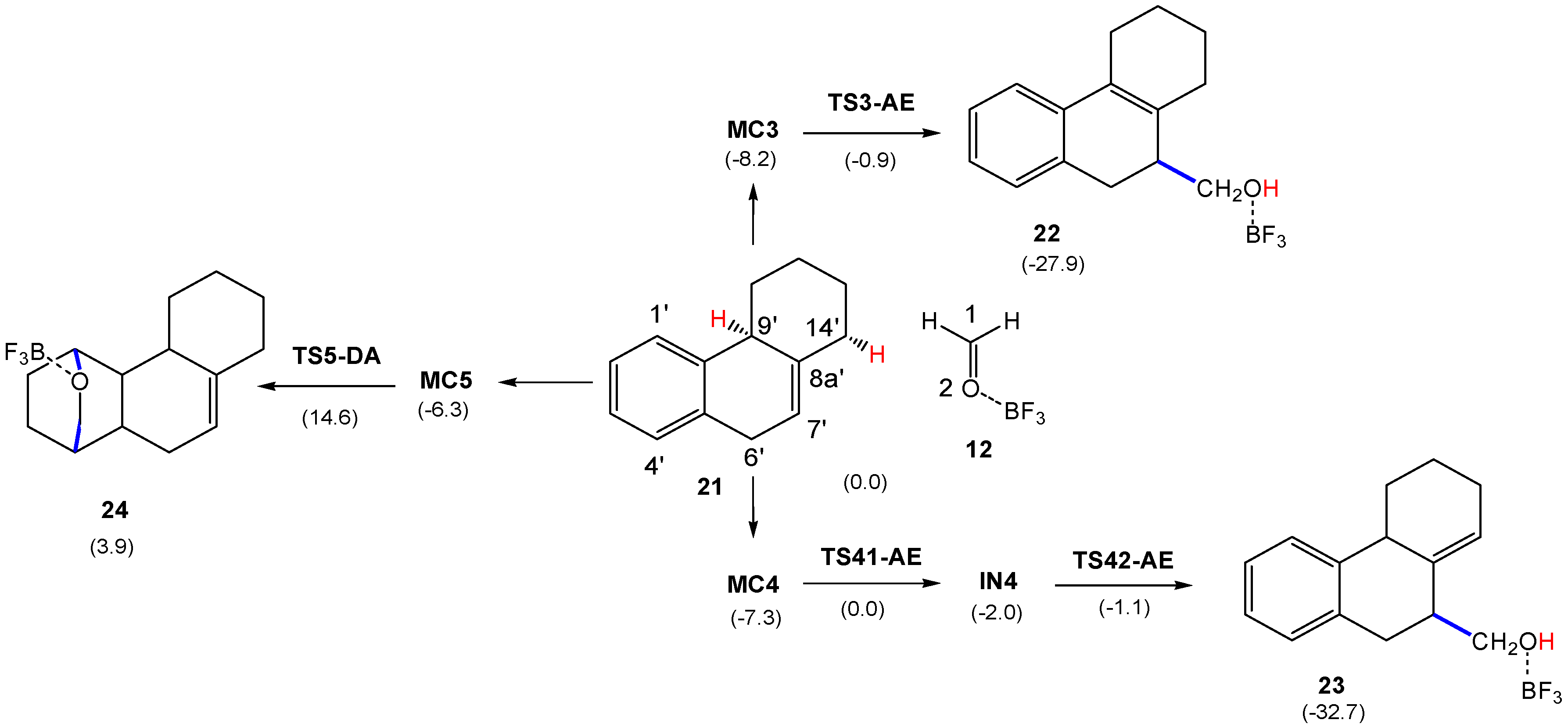
3.3. Study of the P-AE and P-DA Reactions between HPA 21 and LA Complex 12
3.4. Comparative ELF Analysis of the Competitive Polar P-DA and P-AE Reactions between 2MBD 14 and LA Complex 12. Origin of the Pseudocyclic Selectivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Carruthers, W. Some Modern Methods of Organic Synthesis, 2nd ed.; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon Press: Oxford, UK, 1990. [Google Scholar]
- Alder, K.; Pascher, F.; Schmitz, A. Absorption of maleic acid-anhydride and azodicarbon acid-ester in simple unsaturated carbohydrogens. Information on the substitution processes in the allyl position. Ber. Dtsch. Chem. Ges. 1943, 76, 27–53. [Google Scholar] [CrossRef]
- Mikami, K.; Shimizu, M. Asymmetric ene reactions in organic synthesis. Chem. Rev. 1992, 92, 1021–1050. [Google Scholar] [CrossRef]
- Nahm, S.H.; Cheng, H.M. Transition-state geometry and stereochemistry of the ene reaction between olefins and maleic anhydride. J. Org. Chem. 1986, 51, 5093–5100. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry. Part A: Strucutre and Mechanisms, 5th ed.; Springer: New York, NY, USA, 2007. [Google Scholar]
- Fleming, I. Molecular Orbitals and Organic Chemical Reaction; John Wiley & Sons: West Sussex, UK, 2009. [Google Scholar]
- Woodward, R.B.; Hoffmann, R. The conservation of orbital symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Silvi, B.; Pérez, P. The mysticism of pericyclic reactions: A contemporary rationalisation of organic reactivity based on electron density analysis. Eur. J. Org. Chem. 2018, 2018, 1107–1120. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. Understanding the polar mechanism of the ene reaction. A DFT study. Org. Biomol. Chem. 2014, 12, 7581–7590. [Google Scholar] [CrossRef] [PubMed]
- Arundale, E.; Mikeska, L.A. The olefin-aldehyde condensation. The Prins reaction. Chem. Rev. 1952, 51, 505–555. [Google Scholar] [CrossRef]
- Künzer, H.; Sauer, G.; Wiechert, R. Stereocontrolled derivatization of 3-methoxyestra-1,3,5(10), n-tetraenes via lewis acid promoted prins reactions, (n = 7; 8(9)). Tetrahedron Lett. 1991, 32, 743–746. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent Interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinectics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hehre, M.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. In Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.I.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions—Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab nitio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Claredon Press: Oxford, UK, 1990. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keth, T.; Millam, J. GaussView, Version 3; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. USCF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Sáez, J.A. Unravelling the mechanism of the ketene-imine Staudinger reaction. An ELF quantum topological analysis. RSC Adv. 2015, 5, 37119–37129. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Pérez, P.; Ortega, D.E. Why Do Five-Membered Heterocyclic Compounds Sometimes Not Participate in Polar Diels–Alder Reactions? J. Org. Chem. 2013, 78, 2462–2471. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.A.D.; Vale, M.L.C.; Garcia-Mera, X.; Rodríguez-Borges, J.E. 1,3-versus 1,4-[π4+π2] Cycloadditions between methyl glyoxylate oxime and cyclopentadiene or cyclopentene. Tetrahedron 2012, 68, 1682–1887. [Google Scholar] [CrossRef]
- Rhyman, L.; Ramasami, P.; Joule, J.A.; Sáez, J.A.; Domingo, L.R. Understanding the formation of [3 + 2] and [2 + 4] cycloadducts in the Lewis acid catalysed reaction between methyl glyoxylate oxime and cyclopentadiene: A theoretical study. RSC Adv. 2013, 3, 447–457. [Google Scholar] [CrossRef]
- Houk, K.N.; Luskus, L.J.; Bhacca, N.S. Novel double [6 + 4] cycloaddition of tropone to dimethylfulvene. J. Am. Chem. Soc. 1970, 92, 6392–6394. [Google Scholar] [CrossRef]
- Houk, K.N.; Sims, J.; Watts, C.R.; Luskus, L.J. Origin of reactivity, regioselectivity, and periselectivity in 1,3-dipolar cycloadditions. J. Am. Chem. Soc. 1973, 95, 7301–7315. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| 12 | −5.93 | 6.52 | 2.69 | −0.07 |
| 20 | −4.23 | 6.16 | 1.45 | 1.81 |
| s-trans14 | −3.31 | 5.50 | 0.99 | 3.07 |
| s-cis14 | −3.30 | 5.77 | 0.94 | 2.94 |
| 21 | −2.95 | 6.10 | 0.71 | 3.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels–Alder and Polar Alder-ene Reactions. Molecules 2018, 23, 1913. https://doi.org/10.3390/molecules23081913
Domingo LR, Ríos-Gutiérrez M, Pérez P. A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels–Alder and Polar Alder-ene Reactions. Molecules. 2018; 23(8):1913. https://doi.org/10.3390/molecules23081913
Chicago/Turabian StyleDomingo, Luis R., Mar Ríos-Gutiérrez, and Patricia Pérez. 2018. "A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels–Alder and Polar Alder-ene Reactions" Molecules 23, no. 8: 1913. https://doi.org/10.3390/molecules23081913
APA StyleDomingo, L. R., Ríos-Gutiérrez, M., & Pérez, P. (2018). A Molecular Electron Density Theory Study of the Competitiveness of Polar Diels–Alder and Polar Alder-ene Reactions. Molecules, 23(8), 1913. https://doi.org/10.3390/molecules23081913