A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives


 ,
, 

Abstract
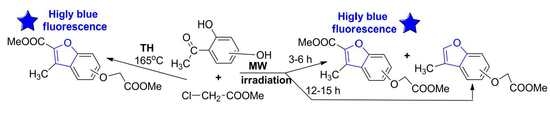
:
1. Introduction
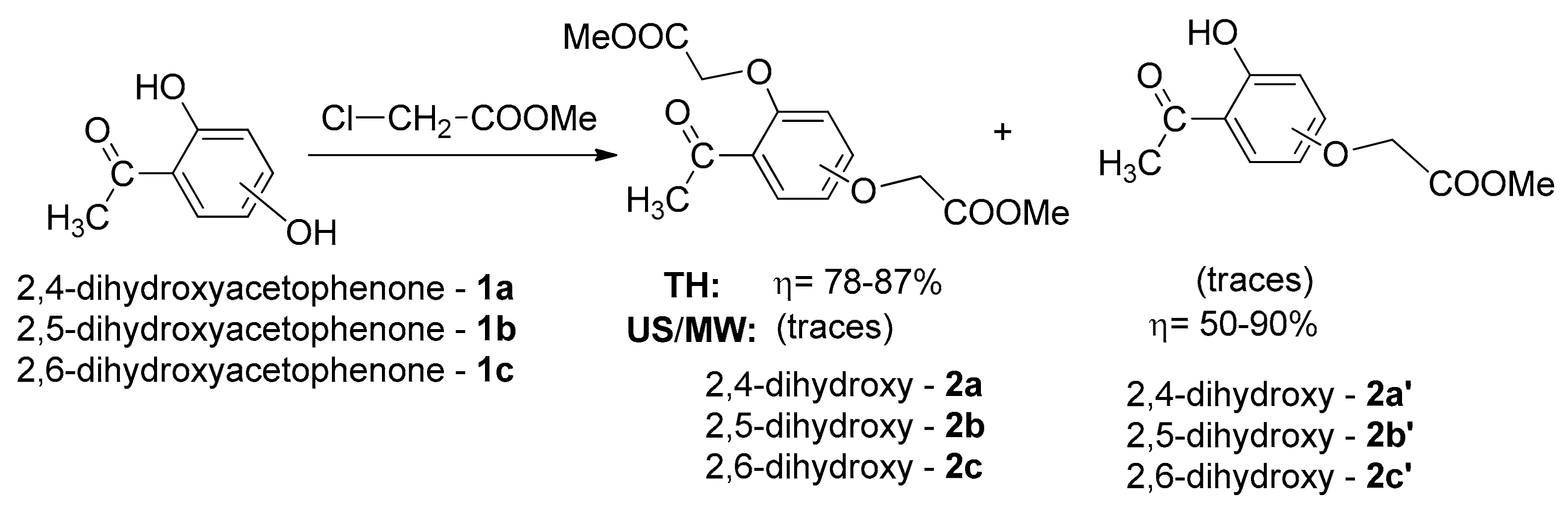
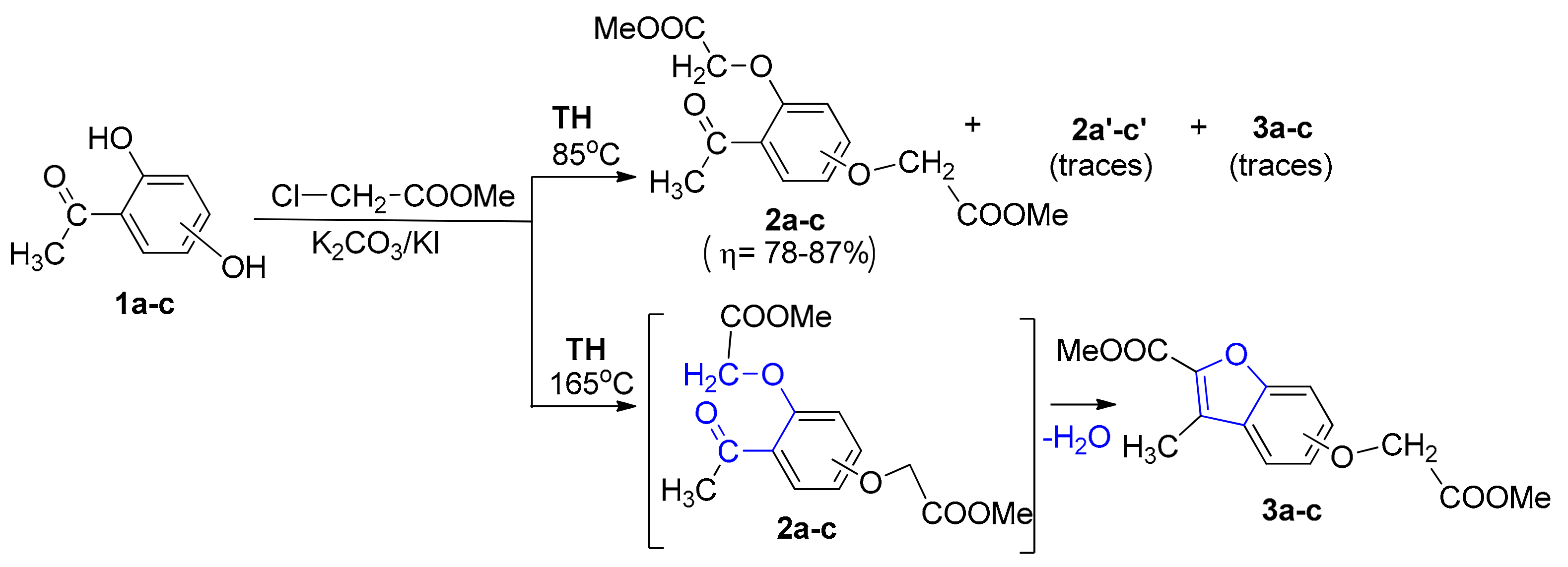
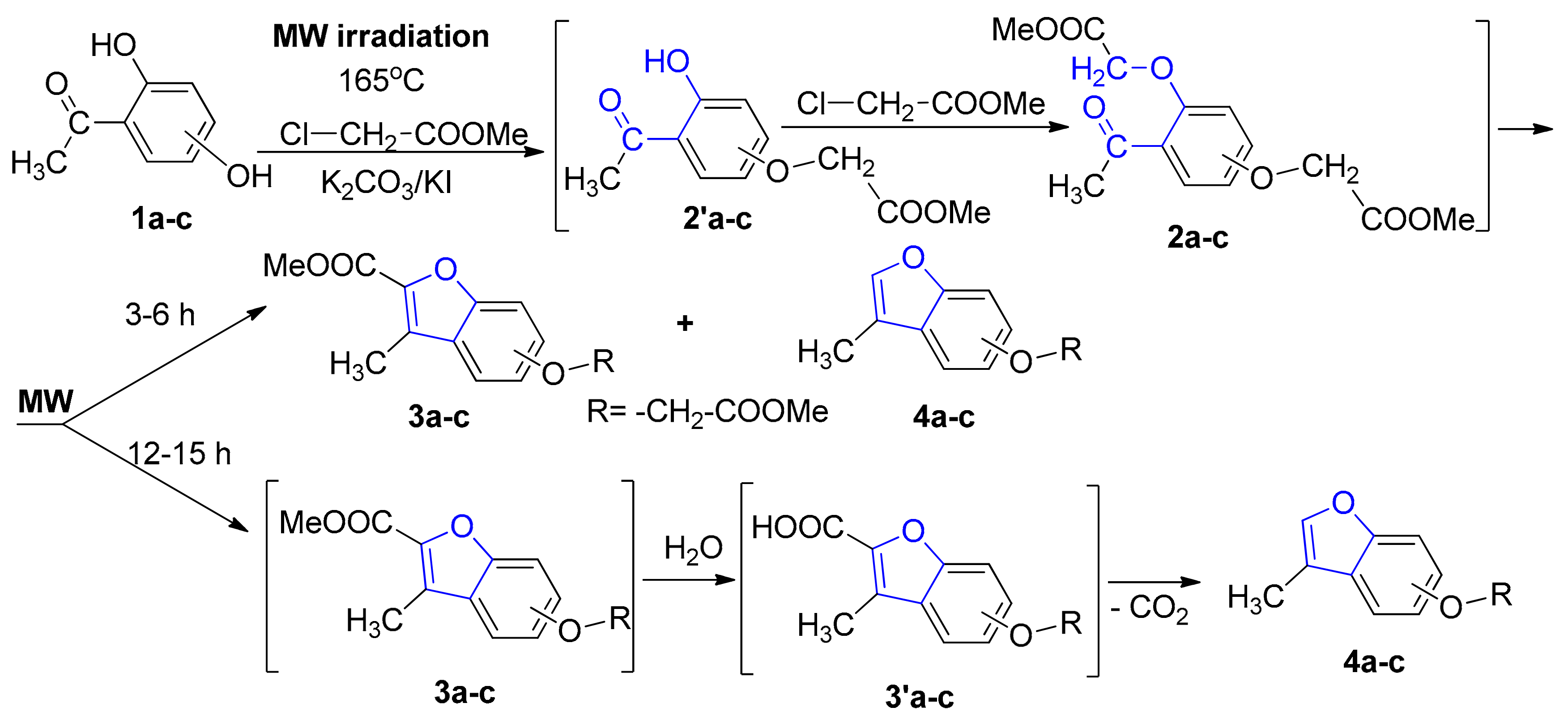
2. Results and Discussion
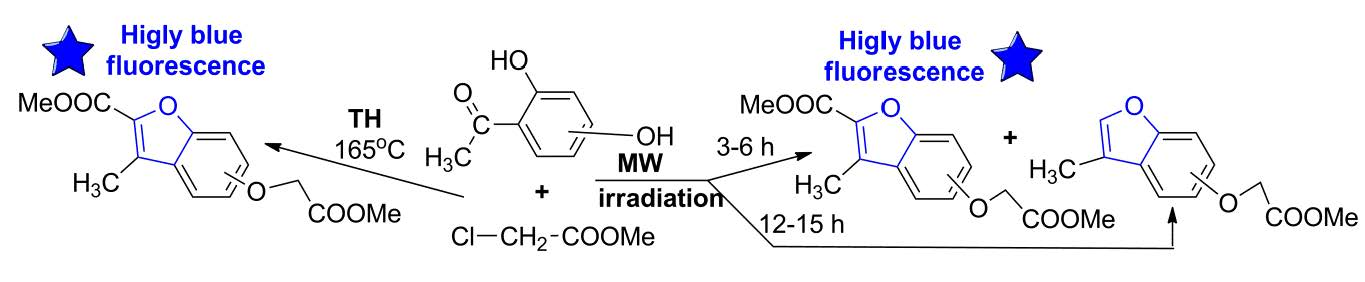
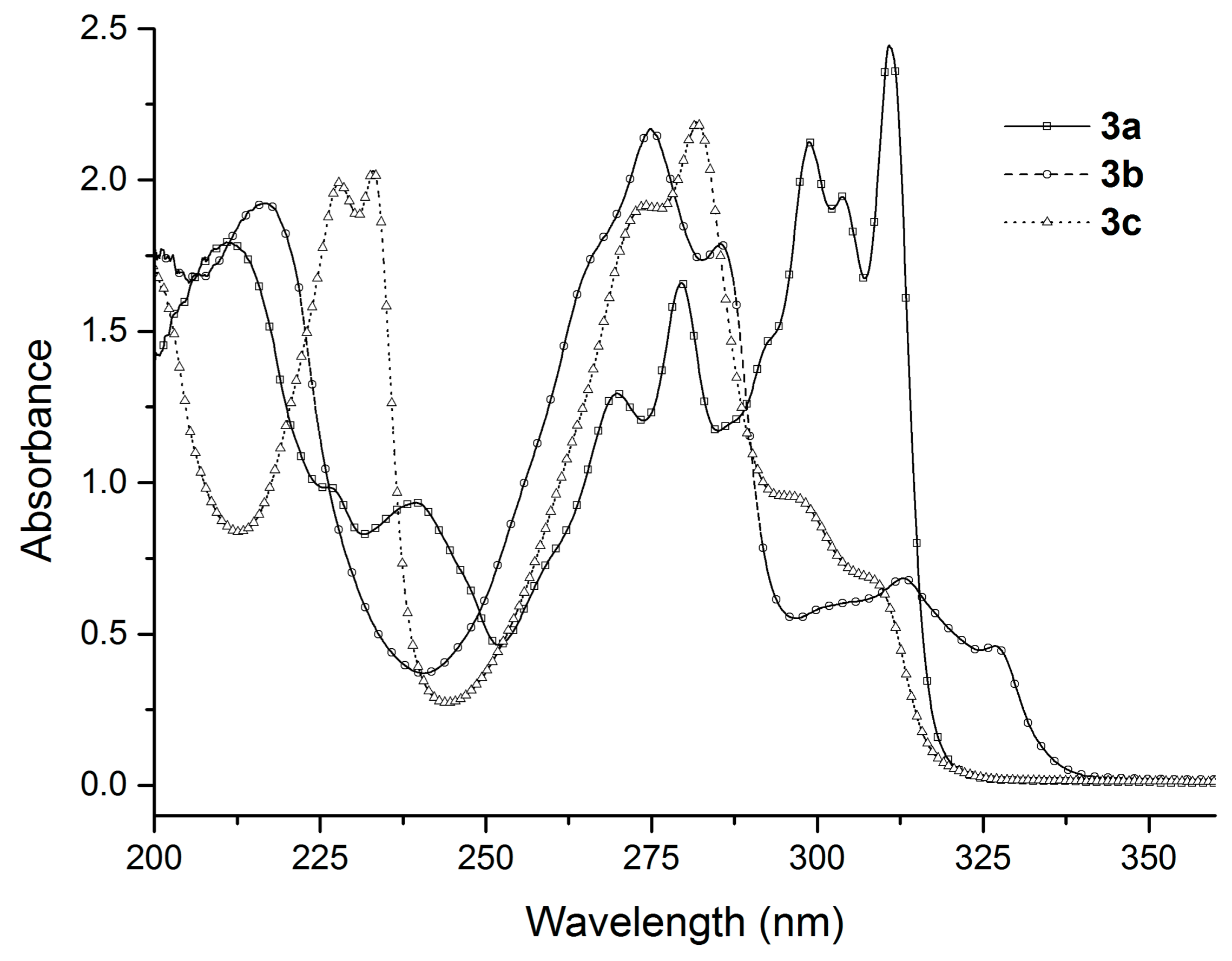
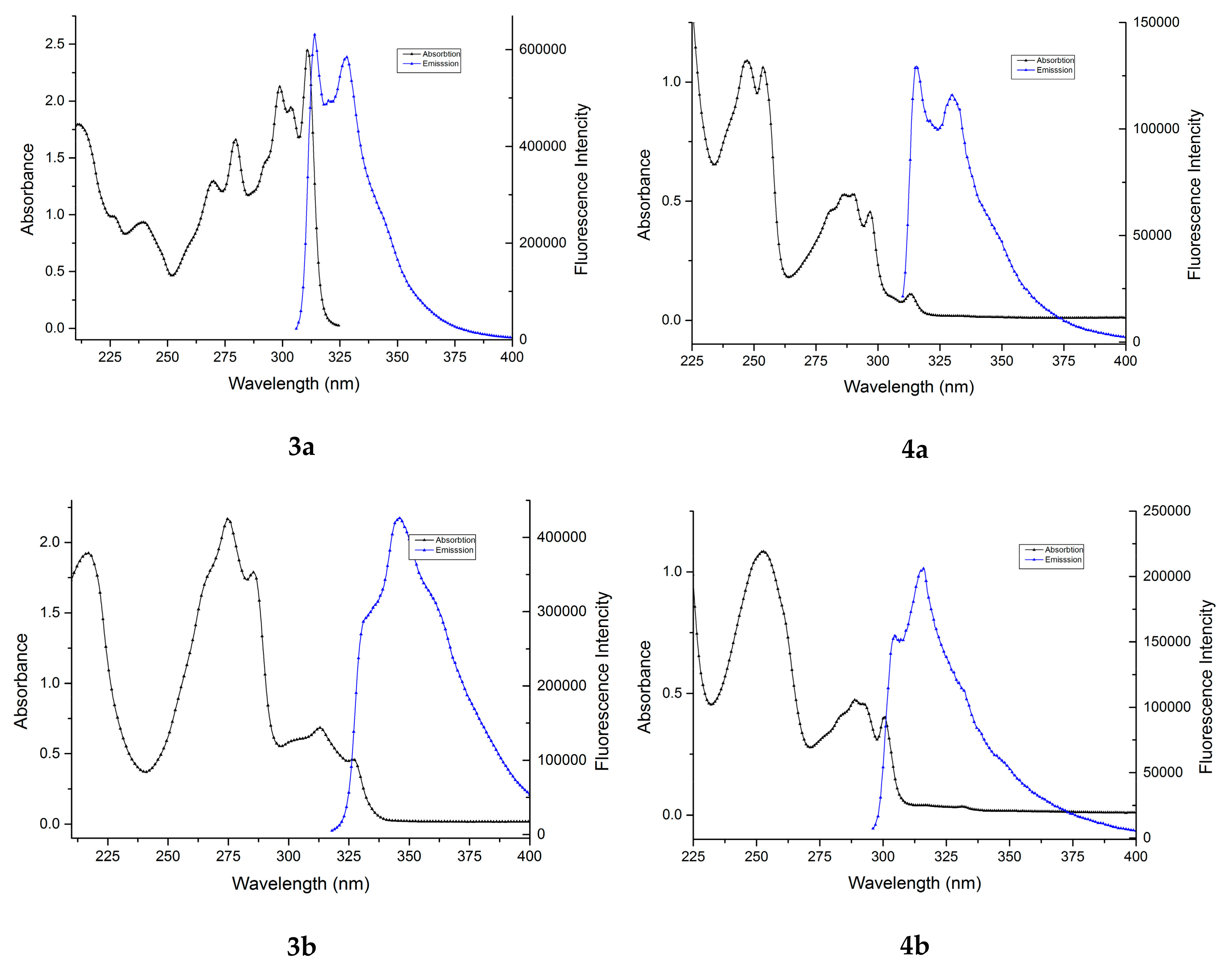
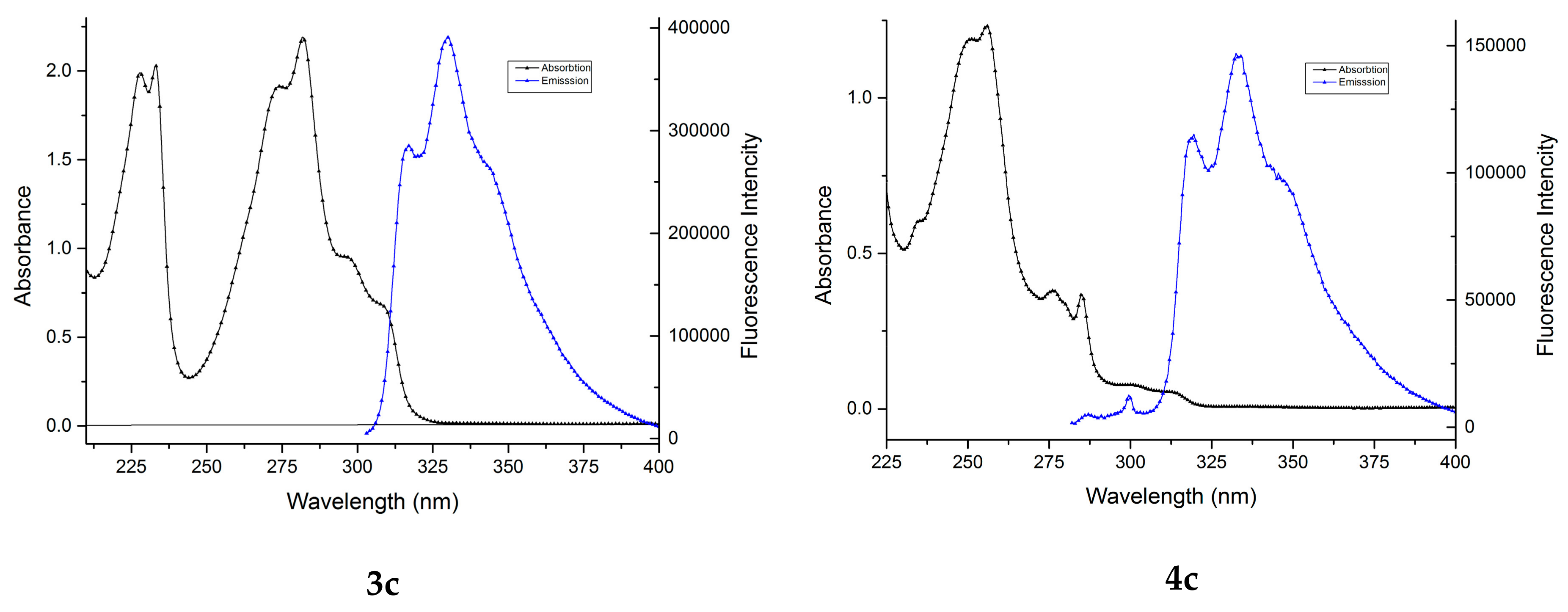
2.1. UV-Vis Study
2.2. Fluorescence Study
3. Experimental Section
3.1. General Procedure
3.1.1. General Procedure for Synthesis of Benzofuran Derivatives 3a–c under Conventional TH Conditions
3.1.2. General Procedure for Syntheses of Benzofuran Derivatives 3a–c and 4a–c under Microwave Irradiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bodke, Y.D.; Shankerrao, S.; Harishkumar, H.N. Synthesis of 2-(1-Benzofuran-2-yl)-4-(1,3-benzoxazol-2-yl/1,3-benzothiazol-2-yl) Quinolines as Blue Green Fluorescent Probes. J. Chem. 2013, 2013, 794810. [Google Scholar] [CrossRef]
- Berlman, B. Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, NY, USA, 1971; ISBN 9780323161671. [Google Scholar]
- Anderson, S.; Taylor, P.N.; Verschoor, G.L.B. Benzofuran Trimers for Organic Electroluminescence. Eur. J. Chem. 2004, 10, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Hwu, J.R.; Chuang, K.S.; Chuang, S.H.; Tsay, S.C. New benzo[b]furans as electroluminescent materials for emitting blue light. Org. Lett. 2005, 7, 1545–1548. [Google Scholar] [CrossRef] [PubMed]
- Kandaz, M.; Güney, O.; Senkal, F.B. Fluorescent chemosensor for Ag (I) based on amplified fluorescence quenching of a new phthalocyanine bearing derivative of benzofuran. Polyhedron 2009, 28, 3110–3114. [Google Scholar] [CrossRef]
- Oter, O.; Ertekin, K.; Kirilmis, C.; Koca, M.; Ahmedzade, M. Characterization of a newly synthesized fluorescent benzofuran derivative and usage as a selective fiber optic sensor for Fe(III). Sens. Actuators B Chem. 2007, 122, 450–456. [Google Scholar] [CrossRef]
- Leung, M.K.; Chang, C.C.; Wu, M.H.; Chuang, K.H.; Lee, J.H.; Shieh, S.J.; Lin, S.C.; Chiu, C.F. 6-N,N-diphenylaminobenzofuran-derived pyran containing fluorescent dyes: A new class of high-brightness red-light-emitting dopants for OLED. Org. Lett. 2006, 8, 2623–2626. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanemura, K.; Horaguchi, T.; Shimizu, T.; Sakakibara, T. Benzofuran derivatives. Part 4, synthesis of benzofurans and 2,3,4,5-tetrahydro-1-benzoxepin-3,5-diones. J. Het. Chem. 1992, 29, 423–429. [Google Scholar] [CrossRef]
- Podea, P.V.; Tosca, M.I.; Paizs, C.; Irimie, F.D. Chemoenzymatic preparation of enantiopure l-benzofuranyl- and l-benzo[b]thiophenyl alanines. Tetrahedron Asymmetry 2008, 19, 500–511. [Google Scholar] [CrossRef]
- Paul, N.M.; Taylor, M.; Kumar, R.; Deschamps, J.R.; Luedtke, R.R.; Newman, A.H. Structure-Activity Relationships for a Novel Series of Dopamine D2-like Receptor Ligands Based on N-Substituted 3-Aryl-8-azabicyclo[3.2.1]octan-3-ol. J. Med. Chem. 2008, 51, 6095–6109. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, T.; Sato, S.; Shibuya, M.; Kanoh, N.; Taniguchi, T.; Monde, K.; Iwabuchi, Y. Enantioselective Intramolecular Aza-Spiroannulation onto Benzofurans Using Chiral Rhodium Catalysis. Heterocycles 2014, 89, 631–639. [Google Scholar] [CrossRef]
- Zeni, G.; Larock, R.C. Synthesis of Heterocycles via Palladium π-Olefin and π-Alkyne Chemistry. Chem. Rev. 2004, 104, 2285–2310. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, T.; Yokoyama, T.; Inamura, K.; Katakura, S.; Komoriya, S.; Yamaguchi, H.; Hara, T.; Iwamoto, M. Dibasic (Amidinoaryl)propanoic Acid Derivatives as Novel Blood Coagulation Factor Xa Inhibitors. J. Med. Chem. 1994, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, F.; Jiang, X.; Zhang, W.; Liu, J.; Liu, W.; Fu, L. Synthesis and antimicrobial evaluation of 3-methanone-6-substituted-benzofuran derivatives. Eur. J. Med. Chem. 2012, 54, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, J.; Kumar, H.V.; Harini, S.T.; Naik, N. Synthesis of benzofuran based 1,3,5-substituted pyrazole derivatives: As a new class of potent antioxidants and antimicrobials--a novel accost to amend biocompatibility. Bioorg. Med. Chem. Lett. 2012, 22, 4773–4777. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Singh, P.; Tewari, A.K. Design, synthesis, docking and anti-inflammatory evaluation of novel series of benzofuran based prodrugs. Bioorg. Med. Chem. Lett. 2014, 24, 2251–2255. [Google Scholar] [CrossRef] [PubMed]
- Telvekar, V.N.; Belubbi, A.; Bairwa, V.K.; Satardekar, K. Novel N’-benzylidene benzofuran-3-carbohydrazide derivatives as antitubercular and antifungal agents. Bioorg. Med. Chem. Lett. 2012, 22, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Filzen, G.F.; Bratton, L.; Cheng, X.M.; Erasga, N.; Geyer, A.; Lee, C.; Lu, G.; Pulaski, J.; Sorenson, R.J.; Unangst, P.C.; et al. Synthesis and SAR of selective benzothiophene, benzofuran, and indole-based peroxisome proliferator-activated receptor delta agonists. Bioorg. Med. Chem. Lett. 2007, 13, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Yang, X.D.; Zeng, X.H.; Xu, X.L.; Zhang, G.L.; Zhang, H.B. Synthesis and cytotoxic activities of novel hybrid compounds of imidazole scaffold-based 2-substituted benzofurans. RSC Adv. 2012, 2, 4612–4615. [Google Scholar] [CrossRef]
- Habermann, J.; Ley, S.V.; Smits, R. Three-step synthesis of an array of substituted benzofurans using polymer-supported reagents. J. Chem. Soc. Perkin Trans. 1999, 1, 2421–2423. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. Nonthermal Effects of Microwaves in Organic Synthesis. In Microwaves in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 134–218. ISBN 9783527314522. [Google Scholar]
- Rodriquez, M.; Taddei, M. Synthesis of Heterocycles via Microwave-Assisted Cycloadditions and Cyclocondensations. In Microwave-Assisted Synthesis of Heterocycle; Van der Eycken, E., Kappe, C.O., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; pp. 213–266. ISBN 9783540309833. [Google Scholar]
- Perreux, L.; Loupy, A.; Petit, A. Nonthermal Effects of Microwaves in Organic Synthesis. In Microwaves in Organic Synthesis, 3rd ed.; de la Hoz, A., Loupy, A., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 127–207. ISBN 9783527331161. [Google Scholar]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbancioc, G.; Zbancioc, A.M.; Mangalagiu, I. Ultrasound and microwave assisted synthesis of dihydroxyacetophenone derivatives with or without 1,2-diazine skeleton. Ultrason. Sonochem. 2014, 21, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Zbancioc, A.M.; Miron, A.; Tuchilus, C.; Rotinberg, P.; Mihai, C.T.; Mangalagiu, I.; Zbancioc, G. Synthesis and in vitro analysis of novel dihydroxyacetophenone derivatives with antimicrobial and antitumor activities. Med. Chem. 2014, 10, 476–483. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Microwaves | Conventional TH | ||||
|---|---|---|---|---|---|---|
| Reaction Time, (Hours) | Temperature, °C | Yield, % | Reaction Time, (Hours) | Temperature, °C | Yield, % | |
| 3a | 5 | 165 | 45 | 96 | 165 | 76 |
| 3b | 6 | 165 | 49 | 96 | 165 | 79 |
| 3c | 3 | 165 | 46 | 96 | 165 | 74 |
| 4a | 5 | 165 | 49 | 96 | 165 | 0 |
| 13 | 88 | |||||
| 4b | 6 | 165 | 46 | 96 | 165 | 0 |
| 15 | 85 | |||||
| 4c | 3 | 165 | 45 | 96 | 165 | 0 |
| 12 | 80 | |||||
| Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 4a | 4b | 4c | ||||||
| λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 | λabs | ε × 103 |
| 211.6 | 17.93 | 216.6 | 19.23 | 227.8 | 19.90 | 247.0 | 10.89 | 252.4 | 10.84 | 235.2 | 6.04 |
| 226.0 | 9.84 | 274.8 | 21.66 | 233.0 | 20.31 | 252.8 | 10.27 | 288.8 | 4.73 | 251.2 | 11.89 |
| 239.4 | 9.33 | 285.4 | 17.87 | 274.2 | 19.15 | 286.4 | 5.27 | 292.0 | 4.57 | 255.8 | 12.32 |
| 269.8 | 12.95 | 313.0 | 6.83 | 281.8 | 21.91 | 290.0 | 5.29 | 300.4 | 4.04 | 276.2 | 3.80 |
| 279.4 | 16.60 | 326.8 | 4.60 | 295.6 | 9.55 | 296.6 | 4.54 | 285.0 | 3.68 | ||
| 298.8 | 21.26 | 313.0 | 1.10 | ||||||||
| 303.8 | 19.44 | ||||||||||
| 310.8 | 24.46 | ||||||||||
| Compound | λmem (nm) | Quantum Yields (%) |
|---|---|---|
| 3a | 314.0 | 49.61 |
| 3b | 345.5 | 54.85 |
| 3c | 330.0 | 41.58 |
| 4a | 315.5 | 18.98 |
| 4b | 316.0 | 20.22 |
| 4c | 333.5 | 17.18 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moldoveanu, C.; Mangalagiu, I.; Isac, D.L.; Airinei, A.; Zbancioc, G. A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules 2018, 23, 1968. https://doi.org/10.3390/molecules23081968
Moldoveanu C, Mangalagiu I, Isac DL, Airinei A, Zbancioc G. A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules. 2018; 23(8):1968. https://doi.org/10.3390/molecules23081968
Chicago/Turabian StyleMoldoveanu, Costel, Ionel Mangalagiu, Dragos Lucian Isac, Anton Airinei, and Gheorghita Zbancioc. 2018. "A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives" Molecules 23, no. 8: 1968. https://doi.org/10.3390/molecules23081968
APA StyleMoldoveanu, C., Mangalagiu, I., Isac, D. L., Airinei, A., & Zbancioc, G. (2018). A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules, 23(8), 1968. https://doi.org/10.3390/molecules23081968